RESEARCH FUNDING AGREEMENT by and between Affimed Therapeutics AG and The Leukemia & Lymphoma Society
Exhibit 10.6
Execution Copy
CONFIDENTIAL TREATMENT REQUESTED UNDER RULE 406 UNDER THE SECURITIES ACT OF 1933, AS AMENDED.
[*****] INDICATES OMITTED MATERIAL THAT IS THE SUBJECT OF A CONFIDENTIAL TREATMENT REQUEST FILED SEPARATELY WITH THE COMMISSION. THE OMITTED MATERIAL HAS BEEN FILED SEPARATELY WITH THE COMMISSION.
by and between
Affimed Therapeutics AG
and
The Leukemia & Lymphoma Society
1
TABLE OF CONTENTS
| 1. | CERTAIN DEFINITIONS. |
5 | ||||
| 2. | AFM13 DEVELOPMENT PROGRAM AND FUNDING. |
12 | ||||
| 2.1 | THE FUNDING DISTRIBUTION AND MATCHED FUNDS. |
12 | ||||
| 2.2 | PAYMENTS. |
12 | ||||
| 2.3 | USE OF FUNDING AND MATCHED FUNDS. |
12 | ||||
| 2.4 | LIMITATIONS. |
12 | ||||
| 2.5 | DONOR DESIGNATED FUNDS. |
13 | ||||
| 2.6 | PRESENTATIONS. |
13 | ||||
| 2.7 | REPORTS; NOTICES. |
14 | ||||
| 2.8 | PROGRAM AUDITS. |
15 | ||||
| 2.9 | COMPETITION. |
15 | ||||
| 2.10 | COMPASSIONATE USE. |
15 | ||||
| 2.11 | PATIENT ASSISTANCE. |
16 | ||||
| 3. | AFM13 RESEARCH ADVISORY COMMITTEE. |
16 | ||||
| 3.1 | AFM13 RESEARCH ADVISORY COMMITTEE: |
16 | ||||
| 3.2 | MEETINGS. |
17 | ||||
| 3.3 | RECOMMENDATIONS. |
17 | ||||
| 3.4 | DURATION. |
17 | ||||
| 4. | CONDUCT OF AFM13 DEVELOPMENT PROGRAM. |
17 | ||||
| 4.1 | RESPONSIBILITY |
17 | ||||
| 4.2 | STANDARD OF CONDUCT. |
18 | ||||
| 5. | REPRESENTATIONS. |
18 | ||||
| 5.1 | MUTUAL REPRESENTATIONS. |
18 | ||||
| 5.2 | AFFIMED REPRESENTATIONS. |
19 | ||||
| 5.3 | DISCLAIMER. |
20 | ||||
| 6. | ADDITIONAL RESEARCH. |
20 | ||||
| 7. | PUBLICATION. |
21 | ||||
| 7.1 | PUBLICATION OF RESULTS. |
21 | ||||
| 7.3 | PUBLICITY; USE OF PARTY’S NAME. |
21 | ||||
| 8. | INTELLECTUAL PROPERTY. |
22 | ||||
| 8.1 | OWNERSHIP. |
22 | ||||
| 8.2 | PREPARATION. |
22 | ||||
| 8.3 | COSTS. |
22 | ||||
| 8.4 | LICENSE TO LLS BACKGROUND INTELLECTUAL PROPERTY. |
22 | ||||
| 8.5 | PATENT PROSECUTION REPORTING. |
23 | ||||
| 8.6 | LLS ASSISTANCE. |
23 | ||||
| 9. | DEVELOPMENT AND COMMERCIALIZATION OF A PRODUCT. |
23 | ||||
| 9.1 | DEVELOPMENT AND COMMERCIALIZATION OF A PRODUCT. |
23 | ||||
| 9.2 | COMMERCIALIZATION OF A PRODUCT. |
23 | ||||
| 9.3 | ROYALTIES. |
24 | ||||
| 9.4 | SALES REPORTS. |
25 | ||||
| 10. | CONFIDENTIALITY. |
26 | ||||
| 10.1 | CONFIDENTIALITY OBLIGATIONS. |
26 | ||||
| 10.2 | EXCEPTIONS TO NON-DISCLOSURE OBLIGATION. |
26 | ||||
| 11. | DISPUTE RESOLUTION. |
27 | ||||
| 11.1 | PROCEDURES MANDATORY. |
27 | ||||
| 11.2 | NEGOTIATION. |
27 | ||||
| 11.3 | MEETING OF SENIOR MANAGEMENT. |
27 | ||||
| 11.4 | FURTHER PROCEEDINGS: |
28 | ||||
| 11.5 | ARBITRATION. |
28 |
2
| 11.6 |
PRESERVATION OF RIGHTS PENDING RESOLUTION. |
28 | ||||
| 11.7 |
STATUTE OF LIMITATIONS. |
29 | ||||
| 11.8 |
FAILURE TO COMPLY WITH DISPUTE RESOLUTION PROCESS. |
29 | ||||
| 12. |
TERM AND TERMINATION; INTERRUPTION. |
29 | ||||
| 12.1 |
TERM. |
29 | ||||
| 12.2 |
TERMINATION FOR BREACH. |
29 | ||||
| 12.3 |
TERMINATION BY AFFIMED OR LLS. |
30 | ||||
| 12.4 |
INTERRUPTION. |
30 | ||||
| 12.5 |
SURVIVAL. |
31 | ||||
| 13. |
INDEMNIFICATION. |
31 | ||||
| 13.1 |
INDEMNIFICATION BY AFFIMED. |
31 | ||||
| 13.2 |
INDEMNIFICATION BY LLS. |
32 | ||||
| 13.3 |
INDEMNIFICATION PROCEDURES. |
32 | ||||
| 13.4 |
INSURANCE. |
33 | ||||
| 13.5 |
LIMITATION ON LIABILITY. |
33 | ||||
| 14. |
MISCELLANEOUS PROVISIONS. |
34 | ||||
| 14.1 |
RELATIONSHIP OF PARTIES. |
34 | ||||
| 14.2 |
GOVERNING LAW. |
34 | ||||
| 14.3 |
COUNTERPARTS. |
34 | ||||
| 14.4 |
BINDING EFFECT. |
34 | ||||
| 14.5 |
ASSIGNMENT AND SUBCONTRACTING. |
34 | ||||
| 14.6 |
ENTIRE AGREEMENT; AMENDMENT AND WAIVER. |
34 | ||||
| 14.7 |
NOTICE. |
35 | ||||
| 14.8 |
SEVERABILITY. |
36 | ||||
| 14.9 |
HEADINGS. |
36 | ||||
| 14.10 |
CONSTRUCTION OF THIS AGREEMENT. |
36 | ||||
| 14.11 |
FURTHER ASSURANCES. |
36 | ||||
| 14.12 |
FORCE MAJEURE. |
36 |
| Exhibit A | – | Budget | ||
| Exhibit B | – | Compound | ||
| Exhibit C | – | Milestones and Payments | ||
| Exhibit D | – | AFM13 Proposal | ||
| Exhibit E | – | Report Schedule |
3

|

| |
| 0000 Xxxxxxxxxx Xxxxxx, Xxxxx 000 Xxxxx Xxxxxx, XX 00000 XXX xxx.xxx.xxx |
Im Xxxxxxxxxxx Xxxx 000 00000 Xxxxxxxxxx, Xxxxxxx xxx.xxxxxxx.xxx | |
This Agreement (the “Agreement”) is made as of August 26th, 2013 (the “Effective Date”), by and between The Leukemia and Lymphoma Society, a New York nonprofit corporation with its principal place of business at 0000 Xxxxxxxxxx Xxxxxx, Xxxxx Xxxxxx, Xxx Xxxx 00000, Xxxxxx Xxxxxx xx Xxxxxxx (“LLS”) and Affimed Therapeutics AG, a German limited liability company with its principal place of business at Xx Xxxxxxxxxxx Xxxx 000, 00000 Xxxxxxxxxx, Xxxxxxx (“Affimed”). LLS and Affimed are sometimes hereinafter referred to individually as the “Party” and together as the “Parties”.
WHEREAS, LLS is a national voluntary health agency which, among other activities, encourages and sponsors research relating to leukemia, lymphoma, Hodgkin’s disease and myeloma (the “Disease”) to develop therapies to cure or mitigate the Disease, and engages in other charitable and educational activities to increase understanding and public awareness of the Disease. To further this mission, LLS provides research funding to entities that can demonstrate after LLS’s review process that their proposed research projects have scientific promise to advance LLS’s effort to find treatments and cures for the Disease and its complications.
WHEREAS, Affimed is in the business of developing pharmaceutical products and has submitted a project proposal and funding request to LLS (the “Affimed Proposal”) and the Affimed Proposal has been conditionally approved by LLS through its Therapy Acceleration Program Committee.
4
NOW, THEREFORE, in consideration of the mutual premises herein contained and for other good and valuable consideration the receipt and sufficiency of which is hereby acknowledged by the Parties, the Parties agree as follows.
1. Certain Definitions.
1.1 “Accounting Standards” means, with respect to LLS, the United States Generally Accepted Accounting Principles, and means, with respect to Affimed, the accounting standards according to the German Commercial Code (HGB), in each case, as generally and consistently applied through the Party’s organization.
1.2 “Affiliate” shall mean, with respect to any Person, any other Person who directly or indirectly, by itself or through one or more intermediaries, controls, or is controlled by, or is under direct or indirect common control with, such Person. The term “control” means the possession, direct or indirect, of the power to direct or cause the direction of the management and policies of a Person, whether through the ownership of voting securities, by contract or otherwise. Control will be presumed if one Person owns, either of record or beneficially, more than fifty percent (50%) of the voting stock of any other Person.
1.3 “Affimed Patents” means any patent right owned or controlled by Affimed or any of its Affiliates existing as of the Effective Date and having patent claims covering the Compound and/or the Product.
1.4 “AFM13 Development Program” means the clinical phase 2a Product development activities based on the Affimed Proposal (as specified in Exhibit D), and certain other activities requested by LLS, including presentations to donors and other interested parties, which have been mutually agreed upon by the Parties, and which shall be conducted by Affimed and funded in part by LLS and which includes the Milestones specified in Exhibit C.
1.5 “AFM13 Research Advisory Committee” or “RAC” means the oversight group described in Section 3.
1.6 “Background Intellectual Property” shall have the meaning set forth in Section 8.4.
1.7 “Budget” shall mean the total budget for the costs and expenses of the AFM13 Development Program agreed to by the Parties and included in this Agreement as Exhibit A, which budget (a) may be amended from time to time solely upon the mutual written agreement of the Parties, and (b) shall detail the projected allocation and use of: (i) the funds to be paid by LLS to Affimed with respect of the Funding; and (ii) the Matched Funds.
5
1.8 “Change of Control Transaction” shall mean (i) the acquisition by another person or entity by means of any transaction or series of related transactions (whether by merger, consolidation or transfer or issuance of capital stock or otherwise) resulting in the transfer of fifty percent (50%) or more of the outstanding voting power of Affimed; or (ii) the sale of assets constituting all or substantially all of the assets of Affimed.
1.9 “Claims” shall have the meaning set forth in Section 13.1.
1.10 “Combination Product” means any Product sold or used in combination with one or more other therapeutically active materials which are not Products.
1.11 “Commercially Reasonable Efforts” shall mean the level of effort, expertise and resources to research, develop, and commercialize a Product where such research, development and commercialization is technically feasible, devoting the same degree of attention and diligence to such efforts that is substantially and materially consistent with industry standards for products at a comparable stage in development, with the objective of achieving First Commercial Sale as soon as commercially practicable.
1.12 “Compassionate Use” shall have the meaning set forth in Section 2.10.
1.13 “Compound” means Affimed’s proprietary compound identified as AFM13, a CD30/CD16A bi-specific Recruit-TandAb, which is described in Exhibit B, including any derivatives thereof.
1.14 “Confidential Information” means the financial terms of this Agreement (other than the amount of the Funding) and any scientific, technical, trade, business or financial information possessed, obtained by, developed for or given to the other Party which is treated by the disclosing Party as confidential or proprietary including, without limitation, Proprietary Material, Development Program Results, research materials and developments, formulations, techniques, methodology, assay systems, formulae, procedures, tests, equipment, data, reports, know-how, sources of supply, patent positioning, relationships with consultants and employees, business plans and business developments, information concerning the existence, scope or activities of any research, development, manufacturing, marketing or other projects of either Party, and any other confidential or proprietary information about or belonging to either Party’s
6
suppliers, licensors, licensees, partners, affiliates, customers, potential customers or others. All information of a confidential or proprietary nature supplied in written, electronic, oral or visual form pursuant to this Agreement shall be considered as being Confidential Information, whether or not marked as such. The following information shall not be treated as Confidential Information: information that, as evidenced by the receiving Party by written records, (a) is in the public domain or is known by others in the Field at the time of disclosure; (b) is in the possession of the receiving Party free of any obligation of confidentiality prior to the time of disclosure as evidenced by written records; (c) subsequently becomes part of the public domain or becomes publicly known through no fault of the receiving Party; (d) subsequently is received by the receiving Party without any obligation of confidentiality from a Third Party who is free to disclose the information; or (e) is independently developed by the receiving Party without the use of any Confidential Information.
1.15 “Development Program Results” means all Program Inventions, data sets, data analyses, reports detailing all optimized conditions and procedures, test results, laboratory notes, techniques, know-how, and any other results that are obtained in the performance of the AFM13 Development Program.
1.16 “FDA” shall have the meaning set forth in Section 2.10.
1.17 “Field” means the treatment of any oncological indications in humans in which an anti CD30+ antibody could be effective, including any and all CD30+ malignancies.
1.18 “First Commercial Sale” means the first sale in an arm’s length transaction for end use of the Product in the Field after receipt of the requisite Regulatory Approval. Sales for test marketing, sampling and promotional uses, clinical trial purposes or compassionate, named patient or similar use shall not be considered to constitute a First Commercial Sale.
1.19 “Funding” shall mean an amount up to, but not to exceed, US$ 4.401 Million, which is to be funded by LLS to Affimed for the AFM13 Development Program in accordance with the terms, and subject to the conditions, set forth in this Agreement.
1.20 “Indemnitee” shall have the meaning set forth in Section 13.1.
1.21 “Intellectual Property Rights” means any and all rights in and to discoveries, concepts, ideas, Proprietary Material, developments, specifications, methods, drawings, designs, flow charts, diagrams, models, formulae, procedures, processes, schematics, specifications,
7
algorithms, apparatus, inventions, know-how, materials, techniques, methodologies, modifications, improvements, works of authorship and data (whether or not protectable under patent, copyright, trade secrecy or similar laws), including patents, utility models, and registered and unregistered designs, including mask works, copyrights, trade secrets, design history, manufacturing documentation, and any other form of protection afforded by law to inventions, models, designs, works of authorship, databases or technical information and applications and registrations with respect thereto.
1.22 “Interruption Payment” shall have the meaning set forth in Section 12.4 (b).
1.23 “Interruption” shall occur if at any time after the Program Termination Date Affimed, its Affiliates, licensees, sublicensees, transferees and/or successors all cease to conduct, or have ceased Commercially Reasonable Efforts with respect to the research, development and commercialization of all Products in the Field for a period of at least *****; provided, however, that (i) if, on or before ***** before the end of such period, Affimed notifies LLS of and explains the reasons for the cessation of Commercially Reasonable Efforts and documents its intention to resume Commercially Reasonable Efforts, the initial ***** period shall be extended to *****; (ii) the extension provided in (i) shall be accorded only once; and (iii) this definition shall not include a Technical Failure.
1.24 “Matched Funds” shall have the meaning set forth in Section 2.1.
1.25 “Milestones” means the agreed upon technical, business or regulatory milestones pertaining to the AFM13 Development Program as outlined in Exhibit C.
1.26 “Net Sales” with respect to any Product shall mean the gross amount invoiced by Affimed and its Affiliates, licensees, sublicensees, transferees and/or successors for Products sold in bona fide, arms-length transactions to Third Parties, less (a) quantity and/or cash discounts from the gross invoice price which are actually allowed or taken; (b) freight, transport, postage, handling and insurance included in the invoice price; (c) amounts repaid or credited by reasons of rejections or return of goods or because of retroactive price reductions specifically identifiable to such Product; (d) amounts payable resulting from government (or agency thereof) mandated rebate programs; (e) Third Party rebates or charge-backs to the extent actually allowed; (f) invoiced customs duties and sales, excise and use taxes (including value-added and
8
similar taxes), if any, actually paid and directly related to the sale that are not reimbursed by the buyer; and (g) any other specifically identifiable amounts included in the Product’s gross invoice price that should be credited for reasons substantially equivalent to those listed above; all as determined in accordance with the selling Party’s usual and customary accounting methods, which are in accordance with Accounting Standards.
In the case of any sale or other disposal for value, such as barter or counter-trade, of any Product, or part thereof, other than in an arm’s length transaction exclusively for money, but excluding any Product provided as samples, for research or for Compassionate Use, Net Sales shall be calculated as above on the value of the consideration received.
In the case of any sale or other disposal of a Product between or among the selling Party and its Affiliates, licensees, sublicensees, transferees and/or successors, for resale, Net Sales shall be calculated as above only on the value charged or invoiced on the first arm’s-length sale thereafter to a Third Party.
In the case of any sale which is not invoiced or is delivered before invoice, Net Sales shall be calculated at the time of shipment or when the Product is paid for, if paid for before shipment or invoice.
In the event the Product is sold as a Combination Product, the Net Sales of the Product, for the purposes of determining royalty payments, shall be determined by multiplying the Net Sales (as defined above in this Section) of the Combination Product by the fraction, A/(A+B) where A is the weighted (by sales volume) average sale price of the Product when sold separately in finished form and B is the weighted average sale price of the other product(s) sold separately in finished form, provided that the formula set forth above shall not apply if the Product is only sold in combination form and if each of the active ingredients in a Combination Product results from the AFM13 Development Program and in each such event the following sentence shall apply: In the event that such average sale price cannot be determined for both the Product and the other product(s) in combination, Net Sales for purposes of determining royalty payments shall be mutually agreed upon in good faith by the Parties based on relative value contributed by each component, which such agreement shall not be unreasonably withheld, conditioned or delayed.
1.27 “Patient Assistance” shall have the meaning set forth in Section 2.11.
9
1.28 “Person” means any individual, corporation, partnership, association, joint-stock company, trust, unincorporated organization or government or political subdivision thereof.
1.29 “Prime Rate” shall mean the average prime rate published in The Wall Street Journal during the relevant period (calculated by dividing (a) the sum of the Prime Rates for each of the days during the relevant period, by (b) the number of days in the relevant period).
1.30 “Product” means any form or dosage of pharmaceutical composition or preparation in finished form labeled and packaged for sale that contains the Compound as an active ingredient (including Combination Products) or a derivative thereof.
1.31 “Program Invention” means any and all new discoveries, concepts, ideas, Proprietary Material, developments, specifications, methods, drawings, designs, flow charts, diagrams, models, formulae, procedures, processes, schematics, specifications, algorithms, apparatus, inventions, know-how, materials, techniques, methodologies, modifications, improvements, works of authorship and data (whether or not protectable under patent, copyright, trade secrecy or similar laws and whether or not patentable or reduced to practice), know-how, materials, methods, models, procedures, processes, schematics, specifications, techniques, tools, and any other forms of technology that are conceived, created, discovered, developed, generated, made or reduced to practice or tangible medium of expression during the performance of this Agreement, whether solely by one or more employees or consultants of Affimed, solely by one or more employees or consultants of LLS, or jointly by one or more employees or consultants of Affimed and one or more employees or consultants of LLS, in each case relating to the AFM13 Development Program and/or the Product, together with all related Intellectual Property Rights.
1.32 “Program Termination Date” shall mean the later of (i) the date when the last Milestone has been paid and (ii) the date when the major activities with respect to the clinical phase 2a study to be performed by Affimed under the AFM13 Development Program have been completed.
1.33 “Proprietary Material” means any and all (i) molecules and/or reagents owned by, licensed to or otherwise proprietary to Affimed, and (ii) derivatives, modifications, improvements, fragments, metabolites, analogs or homologs thereof, which could not have been discovered or made but for the use of Proprietary Materials.
10
1.34 “Regulatory Approval” shall mean, with respect to any country, all authorizations by the appropriate governmental entity or entities necessary for commercial sale of a Product in that country including, without limitation and where applicable, approval of labeling, price, reimbursement and manufacturing. “Regulatory Approval” in the United States shall mean final approval of a new drug application or biologic license application, as the case may be, pursuant to the then-applicable provisions of the Code of Federal Regulations permitting marketing of the Product in interstate commerce in the United States. “Regulatory Approval” in the European Union shall mean final approval of a Marketing Authorization Application, or equivalent.
1.35 “Royalty Cap” shall have the meaning set forth in Section 9.3.
1.36 “Technical Failure” shall mean the inability of the AFM13 Development Program despite the exercise of Commercially Reasonable Efforts to meet the respective Milestones because of (a) material technology/scientific/medical challenges, regulatory hindrances, manufacturing difficulties, or supplier delays that are unlikely to be resolved in a reasonable timeframe; and (b) material unforeseen intellectual property issues that will adversely affect Affimed’s ability to commercialize or market a Product.
1.37 “Territory” shall mean worldwide.
1.38 “Third Party” shall mean any Person which is not a Party or an Affiliate of any Party to this Agreement.
1.39 “Transfer Event” shall have the meaning set forth in Section 9.3.
1.40 “Transfer Payments” shall mean any payments, royalties or other consideration that Affimed or its shareholders actually receives in connection with any licensing or transfer of rights to the Product, including in connection with a Change of Control transaction, other than amounts received from a partner or licensee that are committed to cover future industry standard, fully burdened costs to be incurred by Affimed in the performance of research, development and commercial support activities to be performed by Affimed under a license agreement in connection with a Product. In the event that Xxxxxxx receives non-cash consideration in connection with a license or transfer or in the case of transactions not at arm’s-length, Transfer Payments shall be calculated based on the fair market value of such consideration or transaction, at the time of the transaction, assuming an arm’s-length transaction made in the ordinary course of business.
11
1.41 “Valid Patent Claim” means a patent claim of an issued patent that has not expired or been revoked, held invalid or unenforceable by a patent office, court or other governmental agency of competent jurisdiction in a final and non-appealable judgment (or judgment from which no appeal was taken without the allowable time period).
2. AFM13 Development Program and Funding.
2.1 The Funding Distribution and Matched Funds. LLS agrees to provide the Funding not to exceed US$4.401 million to Affimed to fund the AFM13 Development Program according to the Budget and the Milestones. The Milestones may be revised by agreement of the RAC from time to time, provided that the total amount of the Funding shall not be increased except by an amendment to this Agreement agreed upon by the Parties. Affimed agrees to provide funding for the AFM13 Development Program set forth in the Budget (the “Matched Funds”).
2.2 Payments. All payments to be made hereunder (including, without limitation, pursuant to Section 9) shall be made in United States dollars (“Dollars”).
2.3 Use of Funding and Matched Funds. The Funding and Matched Funds shall be used exclusively for the payment of expenses included in the Budget. Should actual expenses of the AFM13 Development Program funded be less than the expenses included within the Budget, then any excess Funding (after taking into account all committed but not paid or accrued expenditures, reasonably agreed upon by the Parties in good faith) shall be returned to LLS within ***** after the Program Termination Date.
2.4 Limitations. Notwithstanding Section 2.3 above or any contrary provision contained herein, LLS shall not be required to make any payment or additional payment in respect of the Funding:
(a) in excess of US$4.401 million;
(b) upon the occurrence and/or during the continuance of any material default and/or any material breach by Affimed of any of its material covenants or obligations under this Agreement below;
12
(c) if a case or proceeding (i) under the bankruptcy laws of the United States, or relevant non-U.S. law, now or hereafter in effect is filed against Affimed or all or substantially all of its assets and such petition or application is not dismissed within ***** after the date of its filing or Affimed shall file any answer admitting and not contesting such petition, or (ii) under the bankruptcy laws of the United States, or relevant non-U.S. law, now or hereafter in effect or under any insolvency, reorganization, receivership, dissolution or liquidation law or statute of any jurisdiction now or hereafter in effect (whether at law or equity) is filed by Affimed for all or substantially all of its assets; and/or
(d) if this Agreement is terminated by any Party in accordance with Section 12, except in accordance with the section of Section 12 pursuant to which the termination occurs.
2.5 Donor Designated Funds. Where the Funding is, in part or whole, provided by a donor to LLS who requests that the donated funds be restricted for support of Affimed, Affimed agrees as a condition to receiving the Funding to participate in reasonable promotional/publicity activities that do not unreasonably interfere with the AFM13 Development Program and Affimed’s other business activities upon reasonable advance notice, provided, however, that Affimed shall have no obligation to publish or disseminate information that contains Affimed’s Confidential Information or proprietary know-how or trade secrets or will compromise securing patent protection of Xxxxxxx’s Intellectual Property or Project Inventions. Affimed shall be obligated to participate in no more than two (2) such promotional/publicity activities per calendar year. Additional meeting requests shall be discussed and mutually agreed upon by the Parties.
2.6 Presentations. As a condition to receiving the Funding, Xxxxxxx agrees to provide, upon reasonable advance notice by LLS to Affimed, a representative(s) acceptable to LLS for internal and external presentations or meetings regarding the AFM13 Development Program, provided, however, that Affimed shall have no obligation to publish or disseminate information that contains Affimed’s Confidential Information or proprietary know-how or trade secrets or will compromise securing patent protection of Affimed’s Intellectual Property or Project Inventions. Such Affimed representative(s) shall discuss the presentation or meeting with the Team Leaders (as defined in Section 3.1) and designated LLS representatives at least ten (10) days prior to the presentation. Affimed shall acknowledge the support of LLS in all such
13
presentations. Notwithstanding the foregoing, Affimed shall be obligated to participate in no more than two (2) LLS presentations or meetings regarding the AFM13 Development Program per calendar year. Additional presentation requests shall be discussed and mutually agreed upon by both Parties.
2.7 Reports; Notices. Affimed shall with respect to the AFM13 Development Program and Net Sales of Product (x) maintain a system of accounting in accordance with Accounting Standards, (y) keep full and complete financial records and maintain an effective system of internal controls, and (z) furnish to LLS reports and/or notices in accordance with the following and Exhibit E:
(a) Affimed shall provide within ***** prior to each AFM13 RAC meeting a progress report of the AFM13 Development Program since the prior AFM13 RAC meeting.
(b) Affimed shall provide within ***** after the end of each fiscal year ending prior to the Program Termination Date and within ***** after the fiscal quarter in which the Program Termination Date occurs, financial reports which describe the use of the Funding amounts and the Matched Funds (including, without limitation, a detailed breakdown of the actual costs of the AFM13 Development Program and how such Funding amounts and Matched Funds have been allocated and in fact used in respect of the AFM13 Development Program), any Milestones achieved, and a summary of the development activities conducted with respect to Products under the AFM13 Development Program during the applicable fiscal quarter covered by such report, together with such other summary information pertaining to activities in the AFM13 Development Program during such period as LLS may reasonably request in writing, prior to preparation of such report, be included in such report.
(c) Within ***** after the Program Termination Date, a Final Progress Report which shall (i) be prepared by Affimed or an Affimed-approved Third Party, and (ii) set forth a summary of the activities conducted in the AFM13 Development Program and Affimed’s final analysis, summary tables, data listings, results and conclusions from the AFM13 Development Program.
(d) As soon as practicable during the AFM13 Development Program and thereafter, notice of any license, sublicense or transfer of any Program Invention, or subcontract or permitted assignment by Affimed of this Agreement or its rights and/or obligations hereunder, or of any Change of Control Transaction.
14
(e) ***** notice of all material actions, suits, claims, proceedings, investigations and inquiries that directly or indirectly involve or impact the AFM13 Development Program.
(f) ***** and in any event within ***** after January 1 and June 1 of each fiscal year following the Program Termination Date until First Commercial Sale, progress reports and status updates on Affimed’s activities with respect to the Product including, without limitation, the development and/or commercialization of any Products.
2.8 Program Audits. LLS shall have the right (at LLS’s expense, except as provided in this Section 2.8 below), no more than ***** per calendar year, unless the finding of any prior audit warrants audits at more frequent intervals, during normal business hours and upon at least ***** written notice, to have LLS internal audit personnel or a mutually acceptable independent audit firm, that has agreed to comply with the confidentiality requirements contained in this Agreement, to inspect Affimed’s records, as they relate to the AFM13 Development Program to verify that Xxxxxxx has complied with Sections 2.3 and 2.4. In the event that any such examination shows a material misuse of the Funding, Affimed shall pay the cost of the examination and reimburse LLS for the full amount of each such misuse or miscalculation plus interest *****.
2.9 Competition. Subject to the obligations of confidentiality under this Agreement, Xxxxxxx hereby agrees and acknowledges that nothing contained herein shall restrict or prevent LLS’ ability to provide funding to, or take any other action with respect to, any Person that competes with a Product, the business, operations, and/or research of Affimed; and Affimed hereby waives any claim against LLS with respect to any such competing activities.
2.10 Compassionate Use. Prior to Regulatory Approval, if a patient, physician or other Person notifies Affimed or LLS that such Person would like to have established a program to accommodate requests for expanded access and individual patient (including emergency) use, as those terms are used by the U.S. Food and Drug Administration (“FDA”) (collectively, “Compassionate Use”) of the Product, then Affimed agrees to enter with LLS into good-faith discussions about possible ways to provide such Compassionate Use for AFM13.
15
Notwithstanding the preceding sentence, Affimed shall have the authority to make the final decision with respect to any Compassionate Use of the Product. In the event of a Transfer Event, the documents providing for such transfer shall require the transferee to comply with the requirements of this Section 2.10.
2.11 Patient Assistance. After Regulatory Approval, Affimed shall establish a patient assistance program that will allow the patients without access to insurance or other resources to have access to the Product (“Patient Assistance”). LLS shall render assistance to Affimed in the setting up of such Patient Assistance if so required by Affimed. In the event of a Transfer Event, the documents providing for such transfer shall require the transferee to comply with the requirements of this Section 2.11.
3. AFM13 Research Advisory Committee.
3.1 AFM13 Research Advisory Committee: After the execution of this Agreement, all matters concerning the AFM13 Development Program may be monitored and reviewed by the AFM13 Research Advisory Committee, as follows: The AFM13 RAC shall consist of two representatives from each Party. The members of the AFM13 RAC shall have appropriate scientific expertise necessary to monitor the AFM13 Development Program. Each Party may appoint or substitute any of its members serving on the AFM13 RAC by written notice to the other parties. One (1) representative from each Party shall be designated as Team Leader and the Affimed Team Leader shall serve as the Chairperson of the AFM13 RAC. The role of the AFM13 RAC is to review the AFM13 Development Program and to offer advice to Affimed in support of the objectives of the AFM13 Development Program. The AFM13 RAC shall serve the following purposes:
(a) to facilitate communications between the Parties relating to the Product;
(b) to provide advice relating to the AFM13 Development Program and evaluate any proposed revisions to such AFM13 Development Program which may affect the timing or determination of any Milestones;
(c) to review progress toward Milestones and determine whether or not they have been achieved and satisfied, for the purpose of confirming whether and when Milestone payments would be paid;
16
(d) to regularly review the Budget and actual expenditures to ensure that LLS funding is being used by Affimed solely for and in accordance with the Budget;
(e) to determine whether any overrun of the Budget is justified and how such overrun will be funded; and
(f) to review the choice of the assignee, licensee or transferee in connection with any Transfer Event.
3.2 Meetings. The AFM13 RAC shall hold meetings (in person or by teleconference) at such times and places as the Team Leaders may mutually agree, provided that meetings shall be held at least every three (3) months during the AFM13 Development Program, and more frequently if requested by a Team Leader. The first meeting of the AFM13 RAC shall be held within ninety (90) days of the Effective Date. The quorum for AFM13 RAC meetings shall be three (3) members. An Affimed AFM13 RAC member shall keep minutes of the meetings that reflect in reasonable detail all actions recommended or taken. Such minutes shall not be deemed to amend or waive any provisions of this Agreement, and must be reviewed by LLS. Minutes shall be circulated by the Chairperson within ten (10) days after each AFM13 RAC meeting. Either Party shall have the right upon reasonable prior notice to the other to invite non-AFM13 RAC members or external parties/consultants to any AFM13 RAC meeting, provided that any attendee is under confidentiality terms no less stringent than are contained in this Agreement, and mutual agreement that there is no conflict of interest by the external parties/consultants.
3.3 Recommendations. The AFM13 RAC shall be an advisory body, with recommendations rendered by unanimous vote. Implementation of any recommendations of the AFM13 RAC is subject to the reasonable judgment of both Parties.
3.4 Duration. The AFM13 RAC shall remain in existence until the earlier of the Program Termination Date or a Transfer Event.
4. Conduct of AFM13 Development Program.
4.1 Responsibility. Affimed shall have sole responsibility and control over all aspects of the AFM13 Development Program. Without limiting the foregoing, Affimed shall be responsible for management and conduct of the AFM13 Development Program and shall in particular: (a) maintain complete and accurate records of all Development Program Results; (b)
17
provide to the AFM13 RAC a summary of the Development Program Results and other information reasonably requested by the AFM13 RAC for it to monitor progress of the AFM13 Development Program as deemed relevant by the Team Leaders; (c) consider, review and implement amendments or modifications to the AFM13 Development Program from time to time in such manner as may be appropriate based on any interim Development Program Results; and (d) review, substantiate and demonstrate to the AFM13 RAC the accomplishment of Milestones. Without limitation, human subjects studied in the course of the AFM13 Development Program, including a clinical trial conducted by Affimed pursuant to this Agreement shall be the sole responsibility of Affimed and are under no circumstances a responsibility of LLS.
4.2 Standard of Conduct. Affimed agrees to use the Funding solely for the payment or reimbursement of the expenses of the AFM13 Development Program specified in the Budget, and shall use Commercially Reasonable Efforts in its conduct of the AFM13 Development Program to achieve the Milestones, including but not limited to committing, or contracting for, the appropriate staff, laboratories, offices, equipment and other facilities, to conduct the AFM13 Development Program substantially in accordance with the Affimed Proposal. In the event that LLS has a reasonable, good faith basis to believe that Affimed is not using Commercially Reasonable Efforts to achieve the Milestones, LLS shall give written notice thereof to Affimed specifying the basis for such belief and Affimed shall promptly address LLS’ concerns.
5. Representations.
5.1 Mutual Representations. Each Party represents and warrants to the other that (a) it has the power and authority to execute and deliver this Agreement and to perform its obligations set forth in this Agreement; (b) the execution, delivery and performance of this Agreement have been duly and validly authorized and approved; (c) this Agreement is a legal and valid obligation binding of such Party and enforceable in accordance with its terms; the execution, delivery and performance of the Agreement by such Party does not conflict with any agreement, instrument or understanding, oral or written, to which it is a party or by which it is bound, nor violate any law or regulation of any court, governmental body or administrative or other agency having jurisdiction over it; and (d) it shall perform its obligations under this Agreement in accordance with applicable laws, rules and regulations.
18
Without limitation of the foregoing, the Parties warrant that they will comply with (i) the federal anti-kickback statute (42 U.S.C. 1320a-7(b) and the related safe harbor regulations); and (ii) the Limitation on Certain Physician Referrals, also referred to as the “Xxxxx Law” (42 U.S.C. 1395 (n)). Accordingly, no part of any consideration paid hereunder is a prohibited payment for the recommending or arranging for the referral of business or the ordering of items or services; nor are any payments or contributions of free materials intended to induce illegal referrals of business. In the event that any part of this Agreement is determined to violate federal, state, or local laws, rules, or regulations, the Parties agree to negotiate in good faith revisions to the provision or provisions that are in violation. In the event the Parties are unable to agree to new or modified terms as required to bring the entire Agreement into compliance, either Party may terminate this Agreement immediately upon written notice to the other Party; and (ii) the Health Insurance Portability and Accountability Act of 1996 and the regulations promulgated thereunder.
5.2 Affimed Representations. Affimed represents, warrants and covenants to LLS that Affimed, itself or acting through its subcontractors, (a) has the knowledge, skills and experience to perform the AFM13 Development Program, (b) shall obtain and maintain all licenses, permits, consents and other approvals and authorizations required to conduct the AFM13 Development Program and shall do so in conformity with all applicable laws and regulations, and (c) with respect to any Third Party to whom it subcontracts the performance of any aspect of the AFM13 Development Program, it will monitor such subcontractor(s) to insure that they shall obtain all licenses, permits and other approvals and authorizations required to conduct the AFM13 Development Program and shall do so in conformity with all applicable laws and regulations. Affimed shall provide documentation of its and its subcontractor’s licenses, permits, approvals or authorizations at LLS’s reasonable request.
Affimed also represents that it is not debarred and that it does not knowingly use in any capacity, directly or indirectly, the services of any individual or entity which is debarred by the FDA pursuant to 21 U.S.C. Section 335a(a) or (d) for any of the services or research hereunder. Affimed will promptly disclose in writing to LLS if any individual or entity providing services hereunder is debarred or if any action, claim, investigation or legal or administrative proceeding is pending, threatened, (“debarment action”) relating to the debarment of Affimed or any individual/entity performing services upon notice of such debarment action. In the event of debarment or notice of debarment action, LLS shall have the right to terminate this agreement immediately upon written notice to Affimed.
19
Affimed further represents that it is not excluded and does not use in any capacity, directly or indirectly, the services of any individual or entity which is excluded by the Office of the Inspector General (OIG) pursuant to Social Security Act Sections 1128(a), (b) and (c) and or 42 U.S.C. Section 1320a-7 for any of the services or research hereunder. Affimed will promptly disclose in writing to LLS if any individual or entity providing services hereunder is excluded or upon notice of an (“exclusion action”) or if any action, claim, investigation or legal or administrative proceeding is pending and or threatened. In the event of debarment or notice of debarment action LLS shall have the right to terminate this agreement immediately upon written notice to Affimed.
5.3 DISCLAIMER. EXCEPT AS OTHERWISE EXPRESSLY PROVIDED IN THIS AGREEMENT, NEITHER PARTY MAKES ANY WARRANTY, EXPRESSED OR IMPLIED, OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE OF THE AFM13 DEVELOPMENT PROGRAM, DEVELOPMENT PROGRAM RESULTS, OR ANY PRODUCT RESULTING FROM THE AFM13 DEVELOPMENT PROGRAM. The Parties understand and agree that development and commercialization of any Product in the Field will require Regulatory Approval and that no Party is guaranteeing the safety or efficacy of any Product in the Field. The Parties acknowledge that no warranties are being made with respect to the Intellectual Property Rights, nor are any warranties being made with respect to the Development Program Results or Project Inventions. Affimed makes no guarantees as to the success or any outcome of the AFM13 Development Program.
6. Additional Research. The Parties acknowledge a common purpose of developing product(s) useful for the diagnosis, cure or treatment of the Disease and its complications. Achieving this goal may require additional research and development efforts beyond those encompassed within the AFM13 Development Program. The Parties agree to meet no less than ninety (90) days prior to the Program Termination Date in order to (a) evaluate the progress of the AFM13 Development Program, (b) discuss additional research and development opportunities resulting from the AFM13 Development Program, (c) determine any mutual interest in either amending this Agreement and its associated AFM13 Development Program or
20
entering into a new agreement to further such additional research and (d) if mutually agreed upon, the Parties agree to negotiate in good faith any reasonable agreements as may be proposed by either Party within ninety (90) days thereafter.
7. Publication.
7.1 Publication of Results. If either Party determines that scientific findings and results developed in the conduct of the AFM13 Development Program have scientific significance that would be of significant interest to the broader research community, Affimed shall use reasonable efforts to publish or otherwise cause to be publicly disseminated within the research community such scientific findings and results, together with the underlying data, within ***** after the Program Termination Date, provided, however, that Affimed shall have no obligation to publish or disseminate information that contains Confidential Information of Affimed or proprietary know-how or trade secrets or could reasonably be expected to compromise securing patent protection of Project Inventions. Affimed shall acknowledge the support of LLS in all such publications.
7.2 Availability of Materials. Affimed intends to advance the body of general scientific knowledge in the Field by making available the physical materials, research tools and resources developed during or emanating from the AFM13 Development Program that Affimed determines can be made available to academic researchers for non-commercial research, scientific publications, seminar presentations, and publication of patent applications. These materials shall be shared on an “at cost” basis under a Materials Transfer Agreement (“MTA”) executed between a requesting party (“Transferee”) and Affimed or its licensee, provided that the execution of such MTA shall be at Affimed’s or its licensee’s sole discretion. Such MTA shall contain terms customary in the pharmaceutical industry and for transactions of this type.
7.3 Publicity; Use of Party’s Name. Neither Party shall use the name of the other Party, its trademarks, service marks, logos, or the name of any principal investigator, or any employee or agent, for any press release, marketing, advertising, public relations or other purposes without the prior written consent of the other Party, except that (i) within thirty (30) days following execution of this Agreement, Affimed shall be entitled to issue a press release and other public statements in connection with the execution of this Agreement, provided that Affimed has forwarded a draft of such release or statement to LLS prior to publication and has
21
afforded LLS the opportunity to comment on such draft within ten (10) days of receipt thereof, and (ii) either Party may use the name of each other, disclose the existence of this Agreement, and include a general description of the nature of the AFM13 Development Program in any descriptions on its website, in its research portfolio, fundraising activities and its reporting requirements. If Affimed successfully develops the Product, then for a period of at least ***** following the First Commercial Sale, Affimed shall acknowledge LLS’s financial contribution in any announcements or publications made by Affimed directly related to the Product.
8. Intellectual Property.
8.1 Ownership. All inventions made and all data and know-how generated exclusively by either Party or its Affiliates (directly or through others acting on its behalf) prior to and during the term of this Agreement relating to the AFM13 Development Program shall be owned by the Party making the invention or generating the data or know-how claimed, or if such invention is made jointly (a “Joint Invention”), it shall be owned jointly; provided, however, that, LLS hereby grants to Affimed an exclusive worldwide, fully paid up license with the right to sublicense to its rights in any Joint Invention and any invention made by any LLS employee resulting from the AFM13 Development Program for the purposes specified in this Agreement. Notwithstanding the foregoing, any invention made and all data and know-how generated by any members of the AFM13 RAC, whether made individually or jointly, if any, shall be owned by Affimed.
8.2 Preparation. Affimed shall take responsibility for the preparation, filing, prosecution and maintenance of all Affimed Patents, and any patents and patent applications claiming Joint Inventions, and LLS shall use its reasonable efforts to take responsibility for the preparation, filing, prosecution and maintenance of all LLS Patents, if any.
8.3 Costs. Affimed shall be responsible for all costs incurred in the preparation, prosecution and maintenance of Affimed Patents and Joint Inventions.
8.4 License to LLS Background Intellectual Property. If controlled by LLS and necessary for the commercialization of a Product, and to the extent accepted by Affimed, LLS may from time to time grant to Affimed a license for the term of this Agreement, with the right to sublicense, to certain intellectual property of LLS (“LLS Background Intellectual Property”), and certain of such other intellectual property of LLS as may be useful for the development or exploitation of a Product.
22
8.5 Patent Prosecution Reporting. The filing and progress of all patent applications generated to protect Project Inventions filed by Affimed shall be reported in writing by Affimed in a timely fashion to LLS, but no later than ***** prior to the submission date of any document in connection with such patent applications. After receipt of any such report, LLS may request in writing the disclosure of all actions, papers or agreements related thereto, and Affimed shall promptly provide LLS a copy of each such action, paper or agreement within ***** after LLS’s written request. The obligation set forth in this Section 8.5 shall terminate for each patent application upon the issuance of the resulting patent.
8.6 LLS Assistance. LLS will assist Affimed in any reasonable manner in the procurement and maintenance of all Intellectual Property Rights in the Project Inventions, provided, however Affimed shall cover all expense at its sole cost. Without limiting the foregoing, LLS will execute, and cause its employees and representatives to execute, upon Xxxxxxx’s request, any assignments, applications and other documents that Affimed believes may be necessary or appropriate to protect or perfect the Intellectual Property Rights in the Project Inventions. LLS will ensure that its employees and consultants who participate in activities under this Agreement are obligated to assign or otherwise transfer all right, title and interest in and to all Intellectual Property Rights in the Project Inventions to Affimed or its designee and will, as requested by Xxxxxxx, obtain for Affimed the execution of all necessary applications or other documents therefore from any employee or consultant.
9. Development and Commercialization of a Product.
9.1 Development and Commercialization of a Product. Following the completion of the AFM13 Development Program, Affimed intends, at its own expense, to develop, commercialize and bring at least one (1) Product in the Field to First Commercial Sale, including, without limitation, by conducting clinical trials, filing applications for Regulatory Approval, and taking necessary or advisable actions in connection with the manufacturing, marketing, promotion, sales and distribution of the Product in the Field.
9.2 Commercialization of a Product. Affimed and/or its licensees, sublicensees, transferees and successors (as compared to LLS) shall have the exclusive rights to develop,
23
commercialize, market, sell and distribute any or all Products throughout the Territory. Nothing in this Agreement shall be construed to grant to LLS any right or license to any of Affimed’s technology or intellectual property rights and only licenses and rights granted expressly herein shall be of legal force and effect, and no license or other right shall be created hereunder by implication, estoppel or otherwise.
9.3 Royalties. In consideration of LLS’ payments to Affimed and LLS’ licenses to Affimed hereunder, Affimed shall pay to LLS the following:
(a) a royalty equal to ***** of Net Sales from First Commercial Sale until Affimed has paid to LLS in the aggregate royalty payments equal to the Royalty Cap. The “Royalty Cap” means ***** times the amount of the Funding actually provided to Affimed.
(b) after an amount equal to the Royalty Cap has been fully paid in accordance with subparagraphs (a), (c) and (d), a royalty equal to ***** of Net Sales until the earlier of *****
(c) in connection with any transfer of rights to any Product in which the Product constitutes the principal asset being transferred and the granting of any license or option to any Product, ***** of the Transfer Payments in connection with any such transaction (whether such payments are upfront option payments, license fees, milestone payments, or other fees), provided that such transaction occurs up to the time of First Commercial Sale, and further provided that any payment to LLS under this sub-section (c) shall not exceed and shall be credited against the Royalty Cap.
(d) in connection with any Change of Control Transaction, a percentage of the Transfer Payments in connection with any such transaction calculated as follows: ***** provided in each case that such transaction occurs prior to the time of First Commercial Sale; and further provided that any payment to LLS under this subparagraph (d) shall not exceed and shall be credited against the Royalty Cap.
(e) The royalty payments to LLS under subparagraphs (a) and (b) shall be calculated on a Product-by-Product and country-by-country basis and made within *****
24
***** after any calendar quarter in which Net Sales occur and as otherwise provided in subparagraphs (a) and (b). The royalty payments to LLS under subparagraphs (c) and (d) shall be made within ***** of the event giving rise to the royalty.
(f) Affimed shall promptly provide written report to LLS describing any event described in subparagraphs (c) and (d) (a “Transfer Event”) and include with such report a schedule of each such payment that could be received by Affimed as a result of such transaction, and the anticipated timing of such receipt.
9.4 Sales Reports.
(a) Within sixty (60) days after the end of each calendar quarter following the First Commercial Sale, Affimed shall furnish or cause to be furnished to LLS a written sales report or reports covering the relevant calendar quarter setting forth in detail the Net Sales during such period. With respect to sales of Products invoiced in Dollars, the Net Sales amounts and the amounts due to LLS hereunder shall be expressed in Dollars. With respect to sales of Products invoiced in a currency other than Dollars, the Net Sales and amounts due to LLS hereunder shall be expressed in the domestic currency of the party making the sale, together with the Dollar equivalent of the amount payable to LLS, calculated by translating foreign currency sales into Dollars at the exchange rates for the last business day during the relevant period as reported in The Wall Street Journal, Eastern US Edition. If any licensee or sublicensee makes any sales invoiced in a currency other than its domestic currency, the Net Sales shall be converted to its domestic currency in accordance with the licensee’s or sublicensee’s normal accounting principles. Affimed shall keep accurate records in sufficient detail to enable the amounts due hereunder to be determined and to be verified by LLS.
(b) Upon the written request of LLS, at LLS’s expense and not more than ***** in the twelve (12) month period following the receipt by LLS of the report required under Section 9.4(a), unless the finding of any prior audit warrants audits at more frequent intervals, Affimed shall permit an independent accountant selected by LLS and reasonably acceptable to Affimed, to have access during normal business hours to those records of Affimed as may be reasonably necessary to verify the accuracy of the reports furnished by Affimed pursuant to this Section 9.4. LLS shall pay the cost of any such examination, provided, however, that if such examination determines that actual Net Sales were ***** greater than the amount reported by Affimed to LLS, in addition to promptly paying LLS for any additional royalty then due, Affimed shall reimburse LLS its reasonable expenses associated with such examination.
25
(c) In case of any delay in payment by Affimed to LLS not occasioned by force majeure, interest shall be calculated at ***** after the date on which the applicable payment first becomes due from Affimed.
9.5 Royalties Payable to Affimed. In the event that, pursuant to Section 12.4, the Interruption License becomes effective, in lieu of any other royalties pursuant to this Agreement (other than royalties or payments under Section 9.3 previously paid by Affimed to LLS in accordance with this Agreement), the Parties shall share any amount LLS receives with respect to the product as follows: *****. Such royalties shall be paid to Affimed within ***** of LLS receiving any amount giving rise to the royalty payment to Affimed
10. Confidentiality.
10.1 Confidentiality Obligations. For a period of ***** following the last disclosure by a Party of Confidential Information pursuant to this Agreement, the receiving Party agrees that it will maintain the confidentiality of and will not disclose to any Third Party, or use for any purpose other than as contemplated by this Agreement, any Confidential Information furnished to it by the disclosing Party, except as permitted herein. The receiving Party agrees that any dissemination of Confidential Information to its employees shall be limited to the extent reasonably possible and that the receiving Party shall take reasonable steps to instruct all Persons to whom any Confidential Information is disclosed of the confidential nature of such information, the proprietary right of the disclosing Party therein, and the obligation of such person to maintain the confidentiality of such information during and after employment with the receiving Party. The receiving Party shall also take appropriate action to reasonably assure that any consultants, agents or independent contractors of the receiving Party who are hired or engaged by the receiving Party shall comply with the terms of this Section 10.
10.2 Exceptions to Non-Disclosure Obligation. In the event that the receiving Party is required or requested by law or government order to disclose any Confidential Information, the receiving Party will, to the extent permitted by law, (a) promptly notify the disclosing Party of
26
any such request or requirement, and of the circumstances relating to such disclosure and the proposed scope thereof, so that the disclosing Party may seek an appropriate protective order or other appropriate protections, (b) provide reasonable assistance at the disclosing Party’s request so the disclosing Party may seek to obtain a protective order or other reliable assurance that confidential treatment will be accorded the Confidential Information, and (c) disclose only such Confidential Information as is minimally required to be disclosed.
11. Dispute Resolution.
11.1 Procedures Mandatory. The Parties agree that any claim or dispute arising out of or relating to this Agreement, other than breaches of confidentiality obligations, shall be resolved solely by means of the procedures set forth in this Section 11.
11.2 Negotiation. Any Party who wishes to make a claim arising out of or relating to this Agreement must notify the other Party in writing setting forth the claim together with a reasonable description of the facts and circumstances supporting such claim. The Parties have fourteen (14) days after receipt of the claim notice by the other Party to resolve the dispute informally.
11.3 Meeting of Senior Management. If the aforesaid fourteen (14) day period expires without resolution of the claim, either Party may request a meeting between senior management of the Parties to resolve the dispute and shall propose at least three (3) different non-holiday (U.S. or Canadian or German) weekdays (and times) within the ***** after the request when such a meeting may take place, none to be sooner than seven (7) days after the request is received. If none of the times and dates proposed are acceptable to the other Party, that Party shall, not later than ***** after receiving the request, counter-propose in writing at least three (3) different non-holiday weekdays (and times) within the same period, none to be sooner than ***** after the counter-proposal is received. The Party who made the initial request shall respond to any counter-proposed dates in writing not later than ***** after receiving the counter-proposal. Such a meeting may be either by telephone or in person. If a meeting is agreed upon, the Parties must participate unless it is rescheduled by agreement.
27
11.4 Further proceedings:
(a) The Party requesting the meeting may proceed to arbitration if the other Party has not agreed to a meeting or counter-proposed a meeting within ***** after receiving the claiming Party’s request, or has failed to participate in an agreed meeting.
(b) The Party receiving a request for a meeting may proceed to arbitration if the other Party has not agreed to a meeting within ***** after receiving a counter-proposal, or has failed to participate in an agreed meeting.
(c) Either Party may proceed to arbitration if a meeting takes place and the claim is not resolved.
11.5 Arbitration. Any Party entitled under Section 11.4 to proceed with arbitration may submit the claim or dispute to arbitration conducted under the commercial rules of the International Chamber of Commerce (ICC). The arbitration shall be conducted by an arbitrator with relevant experience in transactions comparable to the transactions contemplated by this Agreement, such arbitrator to be appointed in accordance with ICC rules. Such arbitration shall be the exclusive means of proceeding further in the dispute resolution process. The arbitration shall be held in either Frankfurt, Germany or the County of New York in the State of New York, as elected by the moving party, in the English language. The arbitrator is authorized to award such injunctive and monetary relief as he, she or they believe(s) appropriate. The arbitral award shall be in writing, state the reasons for the award, and be final and binding on the Parties. Judgment on the award rendered by the arbitrator may be enforced in any court having competent jurisdiction thereof.
11.6 Preservation of Rights Pending Resolution.
11.6.1 Performance to Continue. Each Party shall continue to perform its obligations under this Agreement pending final resolution of any claim or dispute arising out of or relating to this Agreement unless the Agreement is rightfully terminated or rescinded.
11.6.2 Provisional Remedies. Although the procedures specified in this Section are the sole and exclusive procedures for the resolution of disputes arising out of or relating to this Agreement, either Party may seek a preliminary injunction or other preliminary relief to avoid irreparable harm or to preserve its rights pending resolution of these dispute resolution procedures.
28
11.7 Statute of Limitations. All applicable statutes of limitation and time-based defenses (such as estoppels and laches) concerning a claim subject to this dispute resolution process shall be tolled upon the sending of a notice of such claim as specified in Section 11.4 above, and such toll shall continue until the time ten (10) days after the date that the claimant becomes entitled to commence arbitration hereunder.
11.8 Failure to Comply With Dispute Resolution Process. Any Party may restart the dispute resolution process as to the same claim, but only after either fourteen (14) days have elapsed after the negotiation period set forth in Section 11.2 and no Party has requested a meeting under Section 11.3, or at least one Party becomes entitled to proceed to arbitration under Section 11.5. Upon rightful commencement of an arbitration concerning a claim, any newer dispute resolution process concerning that claim terminates.
12. Term and Termination; Interruption.
12.1 Term. This Agreement shall become effective as of the Effective Date and, unless earlier terminated pursuant to the other provisions of this Section 12, shall terminate at such time as when there are no longer any payment obligations owing from one Party to the other pursuant to Section 9 of this Agreement.
12.2 Termination for Breach. Notwithstanding any provision contained herein or in any other document to the contrary, either Party may, without prejudice to any other remedies available to it at law or in equity, terminate this Agreement upon the occurrence of any of the following events (each a “Default”) (provided, however, that, in each instance (other than pursuant to Section 12.2(d)), the defaulting Party shall have ***** following receipt of written notice from the terminating Party to cure such Default):
(a) Any material violation by the other Party of any applicable law;
(b) Any material breach or default by the other Party in the performance of any of its material covenants or obligations hereunder;
(c) Any representation or warranty made by the other Party in this Agreement that is not true in any material respects; and/or
(d) A case or proceeding (i) under the bankruptcy laws of the United States now or hereafter in effect is filed against the other Party or all or substantially all of its assets and
29
such petition or application is not dismissed within ***** after the date of its filing or the other Party shall file any answer admitting and not contesting such petition, or (ii) under the bankruptcy laws of the United States now or hereafter in effect or under any insolvency, reorganization, receivership, dissolution or liquidation law or statute of any jurisdiction now or hereafter in effect (whether at law or equity) is filed by the other Party for all or substantially all of its assets.
12.3 Termination by Affimed or LLS. Affimed will endeavor to raise the Matched Funds, in accordance with the Budget. If Affimed is unable to raise the Matched Funds within ***** after the Effective Date, either LLS or Affimed shall have the right to terminate this Agreement thereafter by providing written notice to the other. Notwithstanding any other provision of the Agreement, after such notice neither Party shall have any further obligations to the other Party pursuant to this Agreement.
12.4 Interruption.
(a) Interruption License. Affimed hereby grants to LLS an exclusive (even as to Affimed), worldwide, sublicensable license to the Product and any Development Program Results, which license shall be effective in the event of an Interruption (the “Interruption License”). Upon documentation that an Interruption has occurred in accordance with this Section, Affimed shall promptly transfer to LLS a copy of all Development Program Results in the Field. LLS shall notify Affimed in writing if it believes an Interruption has occurred (the “Interruption Notice”). If Affimed disputes the Interruption Notice, it shall respond in writing within ***** of receipt of the Interruption Notice providing specific evidence supporting its response. If LLS disagrees with such response, such dispute shall be resolved in accordance with Section 11 of this Agreement. If Affimed agrees with the Interruption Notice or fails to respond to the Interruption Notice within the specified *****, an Interruption shall be deemed to have occurred. Xxxxxxx has made LLS aware of the fact that a potential investor in Affimed intellectual property has also requested some form of interruption license in the event of a cessation of Commercially Reasonable Efforts relating to the Product. LLS wishes to encourage additional investment in Affimed relating to the Product and accordingly agrees that LLS will engage in good faith discussions with such investor upon Xxxxxxx’s request to determine how the license granted to LLS pursuant to this subparagraph can be reasonably coordinated with a license Affimed may wish to grant to such investor.
30
(b) Interruption Payment. Affimed may elect to pay the Interruption Payment in lieu of the Interruption License by providing notice of such election to LLS and making the Interruption Payment with such notice. The Interruption Payment shall be one (1) time the amount of the Funding actually provided to Affimed plus an annual interest rate of ***** calculated from the date of LLS’ Funding(s) until the payment of the Interruption License. The Interruption Payment shall be applied against the Royalty Cap for purposes of determining any payments to LLS under Section 9.3.
(c) LLS Partnership Accommodations. Notwithstanding subparagraphs (a) and (b), Affimed has expressed its concern that at certain stages of the Development Program the Interruption License could impede Affimed’s efforts to seek a “partner” to commercialize the Product. LLS agrees that it will subsequently discuss any such concerns with Xxxxxxx and a prospective partner at Xxxxxxx’s request and LLS shall make such adjustments to its Interruption rights as LLS determines are reasonably necessary to accommodate such a partnering agreement.
12.5 Survival. The following provisions shall survive the expiration or termination of this Agreement: 7, 8, 10, 11, 12.5, 14.2, 14.9 and 14.10. If LLS terminates this Agreement for reasons of a Default by Affimed pursuant to Section 12.2, then Sections 9 and 12.4 shall also survive such termination. If (i) LLS has performed all of its payment obligations under this Agreement and (ii) Affimed thereafter terminates this Agreement for reasons of a Default by LLS other than a default in payments by LLS, then Section 9.3 shall also survive such termination.
13. Indemnification.
13.1 Indemnification by Affimed. Affimed agrees to indemnify, hold harmless and defend, LLS and LLS directors, officers, representatives, employees and agents and their respective successors, heirs and assigns (each an “Indemnitee”) from and against any and all Third Party claims, losses, expenses, demands, suits, liability or damage for personal injury, property damage or otherwise, including reasonable attorneys’ fees, (collectively “Claims”), arising directly or indirectly from, relating to, or resulting from (a) Affimed’s or any its Affiliates’, sublicensees’ or contractors’ actions in connection with the development,
31
manufacture or commercialization of the Compounds and/or Products, (b) Affimed’s or any of its Affiliates’ negligence or willful misconduct, (c) any material breach of its representations, warranties, covenants or obligations under this Agreement and (d) the conduct of Affimed’s business or operations outside of the AFM13 Development Program.
Notwithstanding the foregoing, Affimed shall have no obligations pursuant to this Agreement to defend or indemnify LLS from any liability, loss, damage or expense to the extent it arises from any of the occurrences listed in Section 13.2 (a) through (d).
13.2 Indemnification by LLS. LLS agrees to indemnify, hold harmless and defend, Affimed and Xxxxxxx’s Indemnitees from and against any and all Claims arising directly or indirectly from, relating to, or resulting from (a) if but only if the Interruption License becomes effective, LLS’ or any its Affiliates’, sublicensees’ or contractors’ actions in connection with the development, manufacture or commercialization of the Compounds and/or Products; (b) any material breach of its representations, warranties, covenants or obligations under this Agreement and (c) the conduct of LLS’ business or operations outside of the AFM13 Development Program.
Notwithstanding the foregoing, LLS shall have no obligations pursuant to this Agreement to defend or indemnify Affimed from any liability, loss, damage or expense to the extent it arises from any of the occurrences listed in Section 13.1 (a) through (c).
13.3 Indemnification Procedures.
13.3.1 In the case of any Claim asserted against an Indemnitee, such Indemnitee shall (i) notify the other Party (the “Indemnitor”) in writing as soon as it becomes aware of any Claim and shall permit the Indemnitor (at the expense of the Indemnitor) to assume defense of any Claim and (ii) cooperate fully with the legal representative chosen by the Indemnitor, who shall be reasonably satisfactory to Indemnitee, provided that the failure of any Indemnitee to give notice as provided herein shall not relieve the Indemnitor of its indemnification obligation hereunder except to the extent that such failure results in a lack of actual notice to the Indemnitor and the Indemnitor is materially prejudiced as a result of such failure to give notice.
13.3.2 Except with the prior written consent of the Indemnitee, such consent not to be unreasonably withheld, conditioned or delayed, the Indemnitor shall not consent to entry of any judgment or enter into any settlement that provides for injunctive or other non-monetary
32
relief affecting the Indemnitee or that does not include as an unconditional term thereof the giving by each claimant or plaintiff to such Indemnitee of a release from all liability with respect to such Claim.
13.3.3 If the Indemnitee in good faith determines, based upon the written advice of outside counsel, that the conduct of the defense of any Claim subject to indemnification under this Agreement or any proposed settlement of any such Claim by the Indemnitor might be expected to affect adversely the Indemnitee’s tax status, reputation, the ability of the Indemnitee to conduct its business or fulfill its mission, the Indemnitee will have the right at all times to take over and assume control over the defense, settlement, negotiations or litigation relating to that portion of the Claim at the sole cost of the Indemnitor (with counsel reasonably satisfactory to the Indemnitor), provided that if the Indemnitee does so take over and assume control, the Indemnitee may not settle such Claim without the written consent of the Indemnitor, such consent not to be unreasonably withheld.
13.4 Insurance. Affimed shall maintain at its own expense, with a reputable insurance carrier, coverage for Affimed, its Affiliates, and their respective employees written on a per occurrence basis commensurate with a reasonable assessment of the risks associated with the research efforts being conducted by Affimed, including, without limitation, comprehensive general liability insurance for claims relating to the performance and lack of performance of Affimed’s obligations under this Agreement and comprehensive general liability insurance for claims for damages arising from bodily injury (including death) and property damages arising out of acts or omissions of a Affimed Party. On or prior to the Effective Date, Affimed shall provide LLS with an insurance certificate from the insurer(s) evidencing each insurance coverage. At its request, LLS may review Affimed’s insurance coverage with relevant Affimed officials from time to time. Maintenance of such insurance coverage will not relieve Affimed of any responsibility under this Agreement for damage in excess of insurance limits or otherwise.
13.5 Limitation on Liability. EXCEPT FOR EACH PARTY’S OBLIGATIONS TO INDEMNIFY A THIRD PARTY PURSUANT TO SECTION 13.1 and 13.2, IT IS AGREED BY THE PARTIES THAT NEITHER PARTY SHALL BE LIABLE FOR ANY SPECIAL, INDIRECT, EXEMPLARY, PUNITIVE OR CONSEQUENTIAL DAMAGES, INCLUDING LOST PROFITS, ARISING OUT OF THIS AGREEMENT OR ITS SUBJECT MATTER.
33
14. Miscellaneous Provisions.
14.1 Relationship of Parties. The Parties do not intend this Agreement to create a legal partnership, joint venture, or agency relationship. There are no third party beneficiaries to this Agreement. The activities and resources of each Party shall be managed by such Party, acting independently and in its individual capacity and the Parties shall have a relationship of independent contractors with respect to each other. Neither Party shall have any express or implied right or authority to assume or create any obligations on behalf or in the name of the other Party or to bind the other Party to any contract, agreement or undertaking with any Third Party.
14.2 Governing Law. This Agreement shall be governed by and construed in accordance with the law of the state of New York, without giving effect to its principles or rules of conflict of laws. The United National Conventions on Contracts for the International Sale of Goods (1980) shall not apply to the interpretation of this Agreement.
14.3 Counterparts. This Agreement may be executed in one or more counterparts, each of which shall be deemed an original, and all of which together shall be deemed to be one and the same instrument. This Agreement may be executed and delivered by facsimile or electronic transmission, which shall be binding on the Party delivering a copy via facsimile or electronic transmission.
14.4 Binding Effect. This Agreement shall be binding upon and inure to the benefit of the Parties and their respective permitted successors and assigns.
14.5 Assignment and Subcontracting. This Agreement may not be assigned by either Party without the prior written consent of the other Party, except that Affimed may assign this Agreement to an affiliate or to a successor in connection with a Change of Control Transaction, or the sale of that portion of Affimed’s assets or business to which this Agreement relates, so long as the affiliate or successor assumes in writing the obligations of this Agreement. Any assignment or attempted assignment in violation of this provision shall be null and void unless agreed upon in writing by both Parties.
14.6 Entire Agreement; Amendment and Waiver. This Agreement and all Exhibits attached hereto, constitute the entire agreement and understanding of the Parties with respect to
34
the subject matter of the Agreement and supersedes any prior and contemporaneous understandings, proposals and agreements, whether written or oral, between the Parties relating to its subject matter. Any amendment, alteration or modification must be in writing and signed by the Parties. Any waiver of any rights or failure to act in a specific instance shall relate only to such instance and shall not be construed as an agreement to waive any rights or fail to act in any other instance, whether or not similar. The AFM13 RAC shall have no right to amend alter, modify or waive any provision of this Agreement.
14.7 Notice. Any notice required or permitted to be given hereunder shall be deemed given: when personally delivered; upon confirmed receipt of electronic delivery by email or facsimile; upon receipt by delivery by recognized overnight delivery service; or five (5) days after being deposited in the mail, with postage prepaid for certified mail, return receipt requested, addressed as follows:
| AFFIMED THERAPEUTICS AG | ||||
| Address: | Im Xxxxxxxxxxx Xxxx 000, 00000 Xxxxxxxxxx, Xxxxxxx | |||
| Senior Management: | Xxx Xxxxx, Ph.D. CEO |
x00 0000 0000 000 x.xxxxx@xxxxxxx.xxx | ||
| For Legal Issues: | Xxxxxxx Xxxxxxx, Ph.D. Chief Financial Officer |
x00 0000 0000 000 x.xxxxxxx@xxxxxxx.xxx | ||
| For Legal Issues, with a copy to: | Freshfields Bruckhaus Xxxxxxxx Attention: Xxxxxx Xxxxxx |
Xxxx Xxxxxxxx 0 00000 Xxxxxxx, Xxxxxxx x00 00 00 00 00 xxxxxx.xxxxxx@xxxxxxxxxxx.xxx | ||
| THE LEUKEMIA & LYMPHOMA SOCIETY | ||||
| Address: | 0000 Xxxxxxxxxx Xxx., Xxxxx 000, Xxxxx Xxxxxx; XX, XX 00000 | |||
| Senior Management: | Xxxxxxx Xxxxxxxx, Ph.D. Senior VP, Research |
000-000-0000 xxxxxxx.xxxxxxxx@xxx.xxx | ||
| For Legal Issues: | Xxxxxxxxx Xxxxxxxx Chief Administrative Officer and Chief Financial Officer |
000-000-0000 xxxxxxxxx.xxxxxxxx@xxx.xxx | ||
| For Legal Issues, with a copy to: | Xxxxxxx & Xxxxxx, PLLC Attention: Xxxxxxx X. Xxxxxxx |
0000 Xxxxxxxxx Xx, Xxxxx 000 Xxxxxxxx, XX 00000 240-482-2848 Xxx@xxxxxxxxxx.xxx | ||
35
or, in each case, to such other address or facsimile number or to the attention of such other person as may be specified in writing by such Party to the other Party.
14.8 Severability. If any provision of this Agreement is inoperative or unenforceable for any reason in any jurisdiction, such circumstances shall not have the effect of rendering the provision in question inoperative or unenforceable in any other case, circumstance or jurisdiction, or of rendering any other provision or provisions herein contained invalid, inoperative, or unenforceable to any extent whatsoever.
14.9 Headings. The headings contained in this Agreement are for purposes of convenience only and shall not affect the meaning or interpretation of this Agreement.
14.10 Construction of this Agreement. In any construction of this Agreement, the Agreement shall not be construed against any Party based upon the identity of the drafter of the Agreement or any provision of it.
14.11 Further Assurances. Each Party agrees to execute all such further instruments and documents and take all such further actions as the other Party may reasonably require in order to effectuate the terms hereof.
14.12 Force Majeure. Neither Party will be in breach hereof by reason of its delay in the performance of or failure to perform any of its obligations hereunder, if that delay or failure is caused by strikes, acts of God or the public enemy, riots, incendiaries, interference by civil or military authorities, compliance with governmental priorities for materials, or any fault beyond its reasonable control. In such event Affimed or LLS, as the case may be, shall immediately notify the other Party of such inability and of the period for which such inability is expected to continue. The Party giving such notice shall thereupon be excused from such of its obligations under this Agreement as it is thereby disabled from performing for so long as it is so disabled. To the extent possible, each Party shall use reasonable efforts to minimize the duration of any force majeure.
IN WITNESS WHEREOF, the Parties have executed this Agreement as of the Effective Date.
36
| AFFIMED THERAPEUTICS AG | ||
| By: | /s/ Xxx Xxxxx | |
| Print Name: | Xxx Xxxxx | |
| Title: | CEO | |
| By: | /s/ Xxxxxxx Xxxxxxx | |
| Print Name: | Xxxxxxx Xxxxxxx | |
| Title: | CFO | |
| THE LEUKEMIA & LYMPHOMA SOCIETY | ||
| By: | /s/ Xxxxxxxxx Xxxxxxxx | |
| Print Name: | Xxxxxxxxx Xxxxxxxx | |
| Title: | Chief Administrative Officer and Chief Financial Officer | |
37
EXHIBIT A
Budget
Budget and Budget Justification
Stage or Phase AFM13
| Start Date: approx. ***** | End Date: approx. ***** | |||||||
| In US $ | Justification | Funding Request | Other Funding (internal or external) |
Total budget | ||||||||||
| FTE costs (Affimed) |
***** | ***** | ***** | |||||||||||
| CMC |
***** | ***** | ***** | |||||||||||
| Clin Phase II |
***** | ***** | ***** | |||||||||||
| Consulting |
***** | ***** | ***** | |||||||||||
| Total |
***** | ***** | ***** | |||||||||||
The costs comprise four categories (FTEs, CMC, clinical and others) totaling in *****. The funding request from LLS is ***** which corresponds to 3,236,288€/4,401,351$. The other ***** of the project costs are funded by Affimed. The material project activities are planned to be executed from *****.
Project structure and process
The project can be segmented in the CMC process and the regulatory clinical process. Both processes are structured in a parallel fashion. As the two main activities overlap to a great extent, the budget is structured in only one phase. CMC process / production and all clinical activities are mainly performed by external service providers. Affimed has established a good and trustful relationship to its partners, so it can be assumed that the already selected and contracted service providers will be at Affimed’s disposal for the continuation of the AFM13 project. However, Affimed’s personnel will be responsible for initiating, monitoring and controlling all activities. Therefore the budget comprises
38
| • | Personnel internal costs |
The FTE costs are calculated with the actual costs of clinical trial assistants, clinical trial managers and management. Further, the charged FTE costs are reasonable *****. The FTE costs are calculated with *****.
| • | CMC process development and production |
In order to apply only the CMC cost per patient, we have segmented clinical and development cost in set up and patient specific (variable) costs. *****.
| Total batch cost estimate ***** |
***** | |||
| - Thereof process development |
***** | |||
| - Thereof production material ***** |
***** | |||
| - Thereof fill finish ***** |
***** |
*****
| • | Clinical activities |
The above described synopsis of the AFM13 Phase II study requires additional investigations with regard to diagnostic parameters, treatment time and the number of sites if compared to the original numbers which are based on our current study:
| • | additional diagnostic measures (i.e. PET-Scan, analytics, NK-cell activation marker and other biomarker) compared to AFM13-101; |
| • | the treatment time was increased ***** which leads to higher treatment costs and CRO costs (monitoring, maintenance costs); |
| • | the number of sites will have to be increased ***** |
Furthermore, in consideration of an average “drop-out” rate of around ***** Affimed expects that up to ***** patients have to be recruited in order to achieve the limit of ***** eligible patients. Due to the fact that only Adcetris treated patients will be included in this clinical trial, *****.
| Clinic cost | ||||
| - Set up cost |
***** | |||
| - Cost per patient |
***** | |||
| - # of patients ***** |
||||
| Total clinic cost: |
***** |
39
| • | Consulting |
The consulting activities are related to external service providers primarily in the field of regulatory activities.
| Total consulting: |
***** |
*****
*****
40
EXHIBIT B
Compound
Description of AFM13
AFM13, also defined as CD30/CD16A, is a tetravalent bispecific, anti-human CD30, antihuman CD16A, recombinant antibody construct based on Affimed’s proprietary TandAb® technology. AFM13 is designed for the treatment of CD30 positive malignancies, i.e. Hodgkin Lymphoma. AFM13 is constructed by means of recombinant technology. The anti-CD30 sequence derived from a murine antibody. The anti-CD16A was isolated out of Affimed’s fully human antibody libraries employing phage display and this antibody is highly specific to the CD16A isoform which is located on NK cells and macrophages and which is known to be responsible for triggering the ADCC. This CD16A antibody does not bind to CD16B.
| Confidential | - 41 - |
AFM13 is produced in mammalian cells as a homodimer and does not contain any Fc parts of a full antibody. The polypeptide comprises four antibody variable domains, each one being separated from the neighboring domains by a ***** linker of ***** amino acids. The four-domain gene product is unable by itself to bind antigen, since the lengths of the linkers between the domains are too short for intramolecular pairing of the corresponding heavy and light chains. However, they are arranged in an order that permits formation of the corresponding antibody VH/VL domains after non-covalent dimerization. The result of this folding process is a homodimeric molecule with a molecular weight of 104 kDa as outlined in figure 1 (see below).

Figure 1: Folding pathway of AFM13. Fv-1 represents the variable domains of the anti-CD16A antibody, both integrated on the outer domains of the AFM13 TandAb, and Fv-2 represents the variable domains of the anti-CD30 antibody, both integrated as the inner domains of the AFM13 TandAb. Fv-1 is human, derived from Affimed’s antibody library and binds specifically to A isoform of CD16. Fv-2 is a murine antibody, derived from Hybridoma HRS3.
| Confidential | - 42 - |
EXHIBIT C
Milestones and Payments
LLS and Affimed agree to the following provisions regarding Milestones and payments in performance of the AFM13 Development Program under the terms of the Agreement.
| Milestone | Milestone Payment | Milestone Event | Projected Date | |||||
| M1 |
$ | * | **** | ***** |
***** | |||
| M2 |
$ | * | **** | ***** |
***** | |||
| M3 |
$ | * | **** | ***** |
***** | |||
| M4 |
$ | * | **** | ***** |
***** | |||
| M5 |
$ | * | **** | ***** |
***** | |||
| M6 |
$ | * | **** | ***** |
***** | |||
| M7 |
$ | * | **** | ***** |
***** | |||
All milestone payments shall become due and payable by LLS within ***** after LLS’ receipt of a written notice from Affimed confirming that the respective milestone event has occurred.
| Confidential | - 43 - |
EXHIBIT D
AFM13 Proposal

|
Therapy Acceleration Program Biotechnology Accelerator Project Proposal Form |
Instructions:
Complete this form in its entirety using the accompanying Guidelines and Instructions document. Failure to provide information may delay the review of the project proposal.
| PRIMARY CONTACT INFORMATION
| ||||
| Last Name
Xxxxxxxx |
First Name
Xxxxx |
Xxxxxx ¨ MD x PhD ¨ Other | ||
|
Title/Role
Director Business Development & Alliance Management | ||||
| Company Name
Affimed Therapeutics AG |
Department / Group | |||||||||||
|
Street Address
Im Neuenheimer Xxxx 582 | ||||||||||||
| City
Heidelberg |
State |
Zip
69120 |
Country
Germany | |||||||||
| Phone
x00 0000 00000 00 |
Fax
x00 0000 00000 00 |
Alternate Phone
x00 0000 00000 00 | ||||||||||
| E-mail Address
x.xxxxxxxx@xxxxxxx.xxx |
Website
xxx.xxxxxxx.xxx | |||||||||||
| Confidential | - 44 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |

|
PROJECT OVERVIEW
Project Title
Phase IIa study of AFM13, a bispecific CD30/CD16A TandAb antibody for the treatment of patients with refractory and/or relapsed Hodgkin Lymphoma
A first in class immunotherapeutic product addressing the high unmet need of a safe and effective therapy for Hodgkin Lymphoma
Project Descriptors
| Disease Diagnostic Group
x Lymphoma ¨ Leukemia ¨ Myeloma ¨ Other ¨ Not Assignable
|
| Specific Disease (if assignable)
|
|
Hodgkin Lymphoma and other CD30 positive malignancies, such as ALCL, CTCL
|
| Technology
|
| ¨ Small Molecule Therapeutic x Biological Therapeutics ¨ Device/Diagnostic
¨ Delivery Technology ¨ Medical Device ¨ Other |
|
Current Stage of Project |
|
AFM13, a CD30/CD16A bispecific RECRUIT-TandAb antibody, is currently being investigated in a first-in-man clinical study (AFM13-101). In the dose escalation Phase I study, a total of 28 patients have been treated and AFM13 has been safe and well tolerated on all dose levels tested. Furthermore, AFM13 showed clear signs of efficacy with an apparent threshold effect at 1.5 mg/kg when applied four times as an infusion at weekly intervals. 8/13 (61.5%) patients treated at this dose level or higher experienced a loss of tumor mass with 3/13 (23.1%) patients having >50% tumor mass reduction. The dose escalation part of the study has been finished without reaching a Maximum Tolerated Dose (MTD). The highest dose of 7mg/kg has been defined as the Maximum Feasible Single Dose (MFSD). Currently, a twice-weekly dosing regimen is being tested at a fixed dose of 4.5mg/kg.
AFM13 has the potential to become a new targeted therapy not only for patients with HL but also for patients suffering from other CD30+ malignancies, such as ALCL (anaplastic large cell lymphoma) and CTCL (cutaneous T cell lymphoma).
|
| Confidential | - 45 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
Target / Pathway / Mechanism of Action
Bispecific antibodies usually do not occur in nature but are constructed by recombinant DNA technologies. The tetravalent bispecific format, as represented by AFM13, is designed to recruit cytotoxic effector cells of the immune system effectively against tumor target cells. TandAbs comprise solely variable domains which are fused via peptide linkers as depicted in the cartoon below.
Natural killer (NK) cells are a potent subset of lymphocytes that can target and lyse tumor cells. In contrast to T lymphocytes, they do not need to be pre-activated. Their inherent cytolytic activity can be induced via the FcgRIIIA receptor (CD16A), which is expressed on the surface of NK cells, macrophages, and activated monocytes.
Hodgkin Lymphoma results from the clonal transformation of cells of B-cell origin, giving rise to pathogenic Xxxx-Xxxxxxxxx cells (RS cells). RS cells are considered to derive from germinal center B cells. RS cells carry the CD30 marker, which is the identifying cell surface antigen of Hodgkin Lymphoma cells.
The recombinant antibody construct AFM13 simultaneously binds to the CD30 antigen on RS cells and to CD16A (FcgRIIIA) on NK cells. This leads to the formation of a so called “immunological synapse”, subsequent activation of NK cells that release cytotoxic substances, granzyme B and perforin, leading to apoptosis and ultimately to lysis of CD30-positive Xxxx-Xxxxxxxxx cells.
Total Funding Requested and Timeframe
The costs comprise four categories (FTEs, CMC, clinical and others) totaling in *****. The funding request from LLS is ***** which corresponds to 3.236 M€ / 4.401 M$. The other ***** of the project costs are funded by Affimed. The material project activities are planned to be executed from ***** until *****.
| Confidential | - 46 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
Summary: (please do not exceed the space provided)
Hodgkin Lymphoma is a fatal disease that can be successfully treated in the majority of cases with chemotherapy and, where necessary, radiotherapy. However, a significant number of patients relapse or become refractory to treatment. Furthermore, the aggressive treatment regimen can give rise to secondary malignant tumors, and the severe side effects frequently prevent elderly patients from receiving or completing the standard treatment. Therefore, there is a high unmet medical need for more selective therapeutics with less side effects for extending and improving the quality of patients’ lives.
Available alternative options for patients who no longer respond to, or are unable to undergo standard chemotherapy/radiotherapy treatment are of limited effectiveness. This is also the case for the antibody drug conjugate Brentuximab vedotin (Adcetris®), which was recently approved for relapsed HL patients. Although showing a high overall response rate, this immunotoxin has a short duration of response of 6.7 months and shows severe side effects. Approximately 90% of patients treated with Brentuximab vedotin required further therapy within less than 1 year.
In view of the urgent need of an effective treatment for HL with a more benign safety profile, Affimed Therapeutics AG has developed the investigational medicinal product AFM13, which is a tetravalent bispecific (CD30/CD16A) recombinant antibody construct (TandAb). The high potency of this molecule results from its ability to specifically recruit cytotoxic immune effector cells (NK cells) for targeting tumor cells (Xxxx-Xxxxxxxxx cells). After the formation of an “immunological synapse” between the killer cell and tumor cell, the NK cells become activated and lyse the Xxxx-Xxxxxxxxx cells via antibody dependent cellular cytotoxicity (ADCC). AFM13 has been extensively characterized in pre-clinical studies. A robust and scalable manufacturing process has been established. The first-in-man study with AFM13 (AFM13-101) started in September 2010 with terminally ill patients. The overall objective of the study AFM13-101 is to determine the safety, tolerability, pharmacokinetics and activity of single cycles of AFM13 in patients with CD30 positive refractory and/or relapsed Hodgkin Lymphoma. A total of 28 patients have been treated: AFM13 was shown to be safe and well tolerated at all dose levels tested so far. Furthermore, AFM13 showed clear signs of efficacy in these poor prognosis patients.8/13 (61.5%) patients treated with 1.5 mg/kg or more of AFM13 applied four times as a short infusion at weekly intervals showed a loss of tumor mass with 3/13 (23.1%) patients having >50% tumor mass reduction. This very encouraging data warrants the further investigation of AFM13 in a phase II study to assess its efficacy, especially in patients with a better prognosis. If AFM13 fulfills expectations, it could become the first immunotherapy for Hodgkin Lymphoma.
In addition to the convincing scientific and medical rationale several unique attributes make AFM13 an interesting drug opportunity: (i) orphan drug status in the US and EU (ii) straight forward development path with a possible BLA (Biologic License Application at FDA) by end of 2017 (iii) combination with standard 1st line therapy to lower toxicity while maintaining efficacy, and finally (iv) limited competition in the growing market of Hodgkin lymphoma and other CD30+ malignancies.
In this proposal Affimed presents its plan for the further clinical development of AFM13 for treating Hodgkin Lymphoma including important decision and evaluation points.
| Confidential | - 47 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
PROJECT PLAN AND BUDGET
Project Title
Phase IIa study of AFM13, a bispecific CD30/CD16A TandAb antibody, for the treatment of patients with refractory and/or relapsed Hodgkin Lymphoma
A first in class immunotherapeutic product addressing the high unmet need of a safe and effective therapy for Hodgkin Lymphoma
Project Plan
I) Introduction to AFM13, Xxxxxxx Lymphoma and methods of treatment
AFM13. AFM13 is a tetravalent, bi-specific TandAb antibody binding to CD30 on Hodgkin tumors and to CD16A (FcgRIIIA) surface receptors on natural killer cells and macrophages to induce cell lysis. A clinical phase 1 trial with AFM13 was initiated at the end of 2010. Data from 28 patients has so far been analyzed. AFM13 is safe and well tolerated and has shown significant activity in the trial. In the highest dose groups (1.5 mg/kg and higher), 3 of the 13 patients showed a tumor reduction of >50% (corresponding to ~23%) and 2 PR and 7 SD were observed in the group as a whole.
Hodgkin Lymphoma and treatment approaches. The American Cancer Society estimates the incidence of Hodgkin Lymphoma to be about 10,491 patients in the US and 6,905 patients in the five major European countries (EU5) in 2009. About 1,350 patients die as a result of the disease per year in the US.
Untreated Hodgkin Lymphoma leads to death in less than a year after diagnosis on average. Fortunately, this disease can be successfully treated in the majority of cases with an aggressive regimen of chemotherapy combined, where necessary, with radiotherapy. Approximately 84% of the patients respond to this treatment, and 48% show a long term remission of up to 15 years. Nevertheless, about 52% of the patients experience a relapse after initial treatment, the majority occurring within the first two years, and about 40% relapse after a second line treatment. Furthermore, the standard-of-care is also associated with severe side effects from both chemotherapy and radiation, which can give rise to secondary malignant tumors, and which frequently prevent elderly patients from receiving or completing the standard treatment. There is therefore a high unmet medical need for therapeutics which have a more selective mechanism of action, and hence an improved side effect profile, in order to extend and improve the quality of patients’ lives.
| Confidential | - 48 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
In order to improve safety and outcome of HL therapy, alternative approaches have been investigated in the clinic comprising antibody-drug-conjugates, monoclonal antibodies and bi-specific constructs.
In August 2012 Brentuximab vedotin (Adcetris®) was marketed for treating HL in the US. This drug is a CD30-directed antibody-drug conjugate for (i) the treatment of patients with Hodgkin lymphoma after failure of autologous stem cell transplant (ASCT) or after failure of at least two prior multi-agent chemotherapy regimens in patients who are not ASCT candidates and (ii) the treatment of patients with systemic anaplastic large cell lymphoma after failure of at least one prior multi-agent chemotherapy regimen. XXX of Brentuximab vedotin in HL patients is 73% and the median duration of response is 6.7 months. It appears to be quite toxic as shown by disorders of the blood, lymph and nervous systems. A significant proportion of patients developed grade 3 and 4 toxicities. In conclusion, although Brentuximab vedotin shows impressive XXX in salvage HL [and ALCL] therapy, its duration of response and its side effect profile are clear weaknesses that need to be addressed by a safer and more durable therapy.
Naked anti-CD30 monoclonal antibody approaches, such as MDX-060 of BMS or SGN-030 of Seattle Genetics (the antibody used in Brentuximab vedotin conjugate), showed disappointing results in phase II clinical studies with a small portion of partial remissions (0-6%) and stable disease. Even antibodies with improved effector mechanisms, such as XmAb-2513 of Xencor, were not able to demonstrate sufficient activity. Currently, none of these antibodies is being tested in further clinical studies.
More promising data was obtained with bispecific antibody constructs. Reagents developed by Biotest and Medarex showed clear efficacy in Phase I/II clinical trial of refractory Hodgkin disease. However, their further developed was discontinued due to manufacturing and scale-up issues.
So far, no immunotherapeutic has been approved for the treatment of Hodgkin Lymphoma or is currently in development. However, based on encouraging clinical results of a bispecific (CD30/CD16) hybridoma antibody produced by Biotest (see above); Affimed developed a novel highly potent tetravalent bispecific antibody, called AFM13.
The development of an immunotherapeutic approach appears to be of great interest for the medical community as indicated by responses to KOL interviews. KOLs in the US and the EU confirmed that AFM13 has potential not only as 3rd line therapy for relapsed and refractory HL patients, *****.
| Confidential | - 49 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
II) AFM13 – Mode of Action in comparison with other immunotherapies and the antibody drug conjugate Brentuximab vedotin
AFM13 Mechanism of action. Bispecific antibodies usually do not occur in nature, they have to be constructed by recombinant DNA technologies. The tetravalent bispecific format, as represented by AFM13, is designed to recruit cytotoxic effector cells of the immune system for targeting tumor cells. Natural killer (NK) cells are a potent subset of lymphocytes that can often recognize and lyse tumor cells. In contrast to T lymphocytes, they do not need to be pre-activated. Their inherent cytolytic activity can be induced via the FcgRIIIA receptor (CD16A), which is expressed on the surface of NK cells, macrophages, and activated monocytes. Hodgkin Lymphoma results from the clonal transformation of cells of B cell origin, giving rise to pathogenic Xxxx-Xxxxxxxxx cells (RS cells) expressingCD30, a cell surface antigen associated with Hodgkin Lymphoma cells.
AFM13 simultaneously binds to the CD30 antigen on RS cells and to CD16A on NK cells. This leads to the formation of a so-called “immunological synapse” with subsequent activation of NK cells that release cytotoxic substances, granzyme B and perforin, leading to apoptosis and ultimately to lysis of CD30-positive RS cells. A sketch of its mechanism of action is shown below
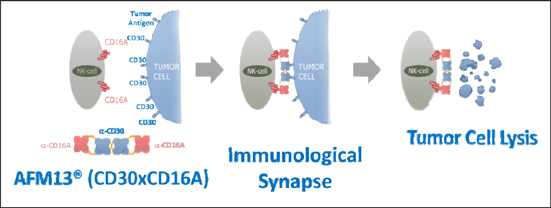
Comparison of AFM13 with other immunotherapies. AFM13 has demonstrated superior ADCC properties on several Hodgkin Lymphoma and Anaplastic Large Cell Lymphoma (ALCL) cell lines relative to an Fc-enhanced anti-CD30 antibody. Its EC50 for tumor cell lysis was 10-fold better. Moreover, in human serum the ADCC of AFM13 was similar to that in standard assay buffers, whereas the activity of the Fc-enhanced antibody was dramatically reduced; the reason being that the anti-CD16A moiety does not compete with polyclonal IgG in human blood and the binding of CD16A by TandAbs is not affected by Fcg receptor polymorphism, unlike existing therapeutic antibodies such as Rituximab. Therefore, better clinical efficacy, lower doses with reduced treatment costs, and fewer side effects to achieve equivalent efficacy can be expected.
| Confidential | - 50 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
Comparison of AFM13 with Brentuximab vedotin. According to publically available data AFM13 has shown a better safety profile than Brentuximab vedotin. Although Brentuximab vedotin is based on an anti-CD30 antibody, its toxicity is primarily due to its chemical payload, the microtubule disrupting agent MMAE (monomethyl auristatin E). It therefore shows a side effect profile similar to other chemotherapeutic agents. In contrast, AFM13 has shown a very benign safety profile in the dose-escalation trial with grade 1 and 2 infusion related side effects.
The mode of action of AFM13 is quite different from that of Brentuximab vedotin. While the latter requires both CD30 internalization and actively dividing Xxxx Xxxxxxxxx cells, AFM13 functions by linking CD30 positive cells with NK-cells. Therefore, CD30 positive Xxxx Xxxxxxxxx cells can be killed without the requirement of CD30 internalization or active cell division.
AFM13’s indiscriminate mode of action makes it an attractive candidate for helping to eradicate minimal residual disease (MRD) and tumors that have become resistant to other treatments. It may therefore also extend the time to relapse. In the ongoing AFM13-101 trial, a total of 9 patients have been enrolled who received Brentuximab vedotin as a prior therapy. 7 of these patients achieved stable disease after AFM13 treatment.
*****, AFM13 should have a significant advantage over Brentuximab vedotin, since it does not increase the amount of toxic substances. ABVD (1st line multi-agent chemotherapy comprising adriamycin, bleomycin, vinblastine and dacarbazine; frequently used in the US) and BEACOPP (1st line multi-agent chemotherapy comprising bleomycin, etoposide, adriamycin, cyclophosphamide, oncovin, procarbazine, prednisone; mainly used in the EU, especially in Germany) have been established 1st and 2nd line therapies for many years. A combination of Brentuximab vedotin with ABVD could not be pursued due to pulmonary toxicity; a combination with AVD is currently being tested.

| Confidential | - 51 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
III) AFM13 – Preclinical Development
Non-clinical pharmacology and pharmacokinetics of AFM13.
Pharmacology. AFM13 binds to human CD16A and to human CD30 with a single digit nanomolar affinity. The in vitro characterization confirmed the specificity of AFM13 for CD16A and revealed comparable binding to both tested CD16A allelic variants (CD16A/V158 vs. CD16A/F158). No binding of AFM13 to different variants of CD16B was detectable. AFM13 induces specific and selective killing of CD30+ target cells by NK cells with an EC50 in the order of 5 – 250 pM (depending on the target cell line and effector cells used). AFM13-induced target cell lysis is specific since AFM13-activated killing of CD30+ target cells by NK cells does not affect CD30- bystander cells. The onset of AFM13-mediated target cell lysis is fast and does not require pre-activation of NK cells. The ability of free soluble AFM13 to induce cytokine release from human PBMC is minimal. Binding of AFM13 to CD16A+ cells in cultures of human PBMC or binding to CD30+ on Hodgkin Lymphoma cells did not give rise to any significant stimulation of proliferation. AFM13 could not be tested with regard to mode of action and efficacy in animals, as no robust and reliable in vivo model based on human tumor and NK cells could be developed.
Pharmacokinetics. Noncompartimental pharmacokinetic analyses in mice and monkeys revealed that AFM13 has linear pharmacokinetics following intravenous administration. Serum concentration time curves in mice and cynomolgus monkeys are biphasic and are strongly dominated by the alpha phase of the kinetics. Only a small portion of the total AUC falls into the beta phase. Terminal half-life of AFM13 in mice is about 6 – 8 h and between 10 and 20 h in cynomolgus monkeys. Some accumulation of AFM13 is seen after repeated administration (q2d x 28d) in cynomolgus monkeys, but effects on clearance and distribution volume are marginal. Due to the dominance of the alpha phase and the rather short alpha half life, substantial steady state levels of AFM13 are not achieved during repeated dosing every other day over 28 days in monkeys.
Toxicology. Cynomolgus monkeys can be regarded as a relevant species for AFM13 from a primary pharmacodynamic perspective. In a dose escalation study evaluating the Maximum Tolerated Dose (MTD) no single dose intravenous MTD could be determined; a single dose of 100 mg/kg of AFM13 was still well tolerated. In a pivotal 28-days toxicity study no overt clinical signs of toxicity were noted throughout all dosing groups with regard to clinical observations, bodyweights, ophthalmoscopy, ECG, blood chemistry, urinalysis, T-dependent antibody response, immunophenotyping, NK cell analysis, organ weights, bone marrow evaluation, macroscopic or microscopic terminal observations. The only finding of note was a dose-dependent decrease in group mean erythroid parameters (RBC, Hb and HCT), with a good reticulocytic response and clear reversibility after two weeks off-treatment. Although leading to borderline anemia at most, this consistent hematological effect of AFM13 was regarded as adverse since longer treatment or higher exposition might possibly lead to symptomatic anaemia. Therefore, the NOAEL was defined as 0.2 mg/kg/dose and was taken as the basis for the determination of a safe starting dose in the first-in-man trials.
| Confidential | - 52 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
Immunotoxicity. No immunotoxic effects of AFM13 could be identified during the 28-day repeat dose toxicity study using the T cell dependent antigen KLH in combination with flow cytometric monitoring of lymphocyte subsets and NK parameters. Furthermore, systemic cytokine release after treatment with AFM13 was absent in all but one animal in the high dose group (this was not rated as drug related). Anti-drug antibody titers in the pivotal 28-day repeat dose toxicity study of AFM13 were negligible and did not impact exposure.
Tissue cross-reactivity. In the human tissues the staining pattern shown by AFM13 was consistent with its specificity, detecting only scattered CD16-positive inflammatory cells in normal tissues. However, AFM13 showed strong immunostaining of a number of secretory-type human epithelia and some non-secretory epithelia. A comparable pattern of epithelial staining has also been observed in cynomolgus monkeys where no toxicity was observed.
IV) AFM13 – GMP manufacturing supplying the clinical development
Manufacturing of Drug Substance (DS). AFM13 is produced *****
Manufacturing of Drug Product – Fill & Finish. *****
Stability Assessment of Drug Substance and Drug Product. ***** The stability of AFM13 is quite impressive for a biological, permitting a shelf-life for AFM13 of at least ***** years.
Manufacturing AFM13 – Summary and Outlook. The established production process appears to be highly robust, and both DS and DP are very stable.
| Confidential | - 53 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
To supply sufficient Drug Product for the upcoming clinical trial and early commercial/market needs AFM13 will be fermented on a *****.
In addition to the established manufacturing process for AFM13 Affimed is currently evaluating several concepts to develop a fully suitable commercial process for the long term market supply after BLA. After discussions and pre-negotiations with CMOs it seems that COGS in the range of *****.
V) AFM13 – Status of clinical development
Phase I study AFM13-101. The First in Human clinical study for AFM13, study code AFM13-101, is being conducted with terminally ill Hodgkin lymphoma patients who progressed or were refractory to previous standard therapy. The study represents an open, single arm, phase I dose escalation trial with the overall objective to evaluate the safety, tolerability, pharmacokinetics, immunogenicity, antitumor activity, maximum tolerated dose (MTD) and the optimal biological dose (OBD) of AFM13 monotherapy. Each patient received a single cycle, consisting of four weekly doses of AFM13. The doses were escalated in cohorts of 3 patients at dose levels of 0.01, 0.04, 0.15, 0.5, 1.5, 4.5 and 7.0 mg/kg. In responding patients, a second course of AFM13 could be administered at the discretion of the investigator. Safety was assessed using CTCAE v. 4.02 criteria. Antitumor activity was assessed by CT and FDG-PET imaging using Cheson criteria. Additionally, a semi-quantitative FDG-PET parameter (maximum standardized uptake value, SUVmax) was used to assess the metabolic activity in targeted lesions after AFM13 therapy. The correlation between the biologic activity of AFM13 and biomarkers (e.g., the plasma concentration of shed CD30 antigen, sCD30) was assessed, as well as the pharmacokinetic profile of AFM13. A total of 28 patients (status early March 2013) have been treated with AFM13: 24 patients in the dose escalation part of the study on a weekly schedule, and 4 patients on the 4.5mg/kg twice-weekly schedule.
| Confidential | - 54 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
Patient characteristic. Patients qualified for study participation if they had active disease at the time of enrolment and failed or progressed on prior standard therapies. The standard therapies in Hodgkin Lymphoma are chemotherapy in first-line treatment (e.g. ABVD or BEACOPP regimen) and high dose chemotherapy plus autologous stem cell transplantation (if patient qualifies) in second line. Patients median age was 38 (range 19 to 72). 22/28 patients had received prior autologous stem cell transplantation and 9/28 patients received prior treatment with Brentuximab vedotin. The median number of prior therapies was 6 (range 3 to 11) overall. 14/28 patients were refractory to their most recent therapy.
Safety assessment. The study confirmed good safety and tolerability profiles for AFM13. All doses from 0.01mg/kg to 7mg/kg once a week, and 4.5 mg/kg b.i.w. proved to be well tolerated and safe. The MTD was not reached. No trend for increased cytokine release has been observed after AFM13 treatment.
25 Patients have been included into the second Development Safety Update Report (DSUR) which was submitted in October 2012. Six SAE (serious adverse event) reports with 9 serious adverse events were documented during the period under review for this DSUR. None of the events was considered possibly related to AFM13. Most frequently reported serious adverse events belonged to the System Organ Class (SOC) “Infections and infestations” (n=7). The following most common non-serious adverse events were identified: fever (n=27), chills (n=16), and headache (n=11). They were mild or moderate in nature and resolved either spontaneously or upon symptomatic treatment/ temporary interruption of AFM13. Fever, chills and headache were identified as symptoms of possible infusion related reactions to AFM13.
Two SUSAR cases were reported during the study. One patient, who was on a dose of 0.5mg/kg and who had known liver involvement, developed a possibly drug-related dose-limiting grade 4 hemolytic anemia and a fatal, not drug-related suspected Aspergillus pneumonia. The other patient, on 4.5mg/kg b.i.w., developed a pneumocystis carinii pneumonia which was judged as being possibly drug-related. The patient fully recovered after antifungal therapy.
It was concluded that hemolytic anemia/reduction in erythroid parameters and infusion related reactions are potential risks associated with AFM13 treatment and should be closely monitored. No other potential risks have been identified for AFM13.
Activity assessment. From 28 treated patients, 26 were eligible for activity assessment. The overall clinical activity of AFM13 in the 26 heavily pretreated patients was 2 partial responses (PR) and 14 stable diseases (SD). Of note, 7 patients who achieved SD have previously been treated with Brentuximab vedotin. 12/26 patients showed a reduction in tumor volume as determined by changes in the Sum of Perpendicular Diameters (SPD) in CT measurement (3 patients > 50%). 14/26 patients showed a reduction in tumor metabolic activity as determined by changes in the SUVmax (3 patients > 50%). The change in the sum of the perpendicular diameters correlated statistically significant to the decrease in the FDG uptake. Activity assessment indicates a threshold effect at 1.5 mg/kg: The treatment of 13 patients with this or higher doses resulted in 2 partial responses and 7 stable diseases.
| Confidential | - 55 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
In alignment, biomarker analysis showed a statistically significant, dose-dependent increase in activated NK cells (as determined by expression of the activation marker CD69), and a reduction in soluble CD30 antigen (sCD30) in plasma. The geometric mean apparent terminal half-life of AFM13 was up to 22.4h, with individual estimates up to 32.6h.
In conclusion, this encouraging data warrants the further investigation of AFM13 in phase II to assess its efficacy in patients with a better prognosis.
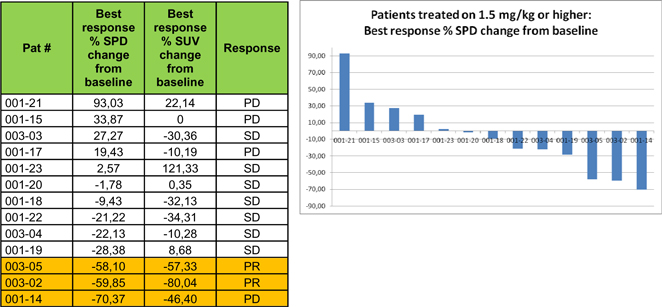
VI) AFM13 – Further clinical development
Planned Phase IIa study to investigate the efficacy of two different dose regimens of AFM13 in Hodgkin Lymphoma patients who progressed or were refractory to standard therapy including Adcetris.
AFM13 will be investigated in a phase II study, using a modified Xxxxx two-stage design, in HL patients who failed or progressed on the therapy with Adcetris. This design allows for the rapid rejection of ineffective treatment at the end of the first stage and provides an estimation of the success rate with a given precision, at the end of the second stage. For this study a targeted response rate of at least 30%, with a precision (standard error, SE) of 5% for Stage 1 and 10% for Stage 2, has been selected.
| Confidential | - 56 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
There will be two treatment groups in Stage 1. Each group will have 9 randomly allocated HL patients. One group will receive 1.5 mg/kg AFM13 dosed twice a week (b.i.w) for 8 weeks and the other group will receive 3.0 mg/kg of AFM13 dosed three times a week (Monday, Wednesday, Friday [MWF]) for 8 weeks. The tumor responses will be assessed 4-6 weeks after the end of therapy, in accordance with the Cheson criteria. The primary objective of this study will be objective response (OR) rate (PR and CR). The secondary objective will be time to relapse.

| * | SE=10% |
At the end of Stage 1 the number of ORs will be assessed and compared between each group. There must be at least one objective response observed in order for this dosing regimen to be considered for progress into Stage 2. If no objective response is observed, this dosing regimen will be confirmed as ineffective and will not be investigated further. The dosing regimen with more responses or more CRs will progress into Stage 2. If there are an equal number of responses in each group then the 1.5mg/kg twice a week dosing regimen will be selected for Stage 2.
An additional 16 patients will be enrolled into Stage 2. At the end of this study the total response rate and efficacy of AFM13 will be estimated. A total of 40 patients to include 34 eligible patients will complete the study.
Affimed will conduct the Phase IIa study in the US and in the EU. There have already been several promising contacts with potential sites in the US. Most recently ***** confirmed their interest and agreed to act as the leading clinical site in the US. And the *****, which are recruiting for the ongoing Phase I trial, also confirmed their intention to participate in the planned Phase IIa study.
| Confidential | - 57 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
Additional sites in the USA and Germany will be contacted and evaluated. The total number of initiated sites will depend on the final evaluation and the estimated patient recruitment.
Clinical Development Plan to BLA submission [is not part of the LLS funded development]
*****
| * | If Stage 1 shows ³30% PR or CR, then AFM13 Stage 2 can switch into a pivotal, open-label, single arm, accelerated approval study with up to a total of 91 patients. |
Rationale on the Phase IIa study – Decision point at stage 1 with >30% OR
The following aspects have been considered to define ³ 30% OR as the target response rate:
| • | AFM13 phase I data |
Affimed analysis of the Phase I results showed that a ³ 20 % response rate is more than feasible and attainable. Out of 13 patients receiving doses of 1.5 mg/kg and higher, 2 patients achieved a partial response, which corresponds to a response rate of 15% (2/13). One additional patient showed a reduction in SPD of > 50%, however in this patient new lesions were diagnosed after therapy, therefore the response had to be assessed as PD. A biopsy revealed that the new lesions were CD30 negative (conversion of HL to a different lymphoma type, oral communication) and thus could not respond to AFM13 therapy. Taking this into account, 3/13 patients showed a significant response, which corresponds to an overall rate of 23%.
However, the LLS TAP committee and Xxxxxxx had agreed upon defining ³ 30% OR as the target response rate, as this would *****
| Confidential | - 58 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
| • | Patient selection |
In the Phase IIa study Affimed will use more stringent patient inclusion criteria as compared to its Phase I (see below) and will enroll better prognosis patients, e.g. with only 2-4 prior treatments, including Adcetris. In addition, CD30 positivity will be confirmed within 6 months prior to start of the AFM13 therapy. However, all patients that will be recruited in this Phase IIa will be “Adcetris failure” patients. This means that all patients received Adcetris as one of their previous treatment options and were either refractory to Adcetris or relapsed.
Note: Phase I patients had received, on average, 6 prior therapies (ranging from 3-11 prior therapies) and were mainly poor prognosis patients. Nine out of 26 eligible patients progressed or were refractory to Adcetris, eight of which were eligible for the activity assessment. Six of these patients achieved stable disease after the AFM13 treatment; four of them had reduced tumor size and reduced metabolic activity in comparison to their baseline values.
| • | Schedule |
In the phase I study patients were treated for 4 weeks either with a weekly schedule (24 patients) or a 2x/week schedule (4 patients). The analysis of PK and NK cell activation showed that the treatment schedule can be optimized. In the Phase IIa study patients will be treated either with a 2x/week or 3x/week (Monday, Wednesday and Friday) schedule.
Rationale for choosing 1.5 mg/kg twice a week (b.i.w.) and 3 mg/kg 3 times a week for 8 weeks
In the phase I study Affimed tested two different dosing regimens: 0.01 mg/kg to 7 mg/kg once a week for 4 weeks (total of 4 doses), and 4.5 mg/kg twice a week for 4 weeks (total of 8 doses). In the weekly schedule we observed a threshold effect with 1.5mg/kg. The doses that included 1.5 mg/kg, 4.5 mg/kg and 7 mg/kg showed a reduction in the tumor volume (SPD) in 5 out of 9 patients, three of them having a reduction of over 50%. 4 patients have so far completed the cohort of 4.5 mg/kg given twice a week resulting in: 1/4 patients progressed, 2/4 patients have SD and 1/4 had PR.
PK/PD analysis from the phase I study shows that activity is not specifically Cmax or AUC dependent. It appears that a threshold concentration/exposure achieved with 1.5mg/kg is required to elicit activity.
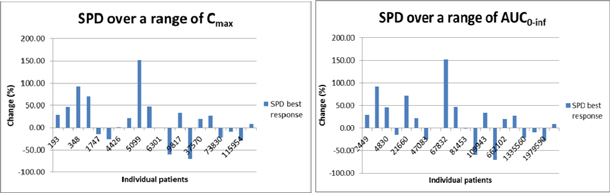
| Confidential | - 59 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
This is consistent with the finding that the amount of activated effector NK cells significantly increases with the dose of AFM13. The data have shown that in the once a week schedule, the amount of activated NK cells in the plasma corresponded to the reduction in the AFM13 plasma concentration. At this stage it is unclear whether the continuation of activity depends on maintaining the plasma concentration of AFM13 above a certain level or whether it requires repeated peaks in its plasma concentrations to maintain the amount of activated NK cells above the threshold level. Further and vice versa it is also not known whether there is an upper AFM13 threshold concentration above which there is saturation of NK cell activation and/or a decrease in the total number of available NK cells in the body.
For the Phase IIa study two additional regimens will be explored. The selection of these two dose schedules is based on the analysis of the data from the Phase I study and the following PK simulations performed. In the table below the simulation of the minimal and maximal steady state concentrations of AFM13 for various regimens given for 4 weeks is shown:
| Cssmin (µg/mL) |
Cssmax (µg/mL) |
|||||||
| 1.5mg/kg 2 times weekly |
0.01 | 11.00 | ||||||
| 1.5mg/kg 3 times weekly |
0.05 | 11.00 | ||||||
| 3mg/kg 3 times weekly* |
0.50 | 37.00 | ||||||
| 4.5mg/kg 2 times weekly |
0.30 | 80.00 | ||||||
| * | As no data are available at 3 mg/kg, simulations were performed based on the models for 1.5 mg/kg and 4.5 mg/kg data. As these models have different parameter estimates (e.g. clearance estimates are 2 fold different), the Cssmin and Cssmax were taken to be between the two simulations. |
In the phase I study the 7 mg/kg dose of AFM13 given once a week achieved very high peak concentrations. This dose also provided a drug presence throughout the period of 7 days. However, it did not result in the expected activity, confirming that a high Cmax on its own did not contribute to the response.
The dose of 4.5 mg/kg given twice a week produced lower peak concentrations than 7 mg/kg, but was present in the blood at a much higher level over the 7 day period. This dose regimen achieved better responses (1PR 2SD and 1PD) than 7 mg/kg (3SD). The fact that 1.5 mg/kg dosed once a week produced relatively good responses (1PR, 1PD – judged on new lesion which was CD30 negative, otherwise it would be PR; and 1 PD) indicates that there must be a fine balance between the dose and the type of exposure (Cmax and Cmin). To explore this we performed pharmacokinetic stimulations in order to assess how different doses and schedules affect the plasma profile of AFM13.
The AFM13 dose of 1.5 mg/kg given once a week in the Phase I study was found to be the lowest dose at which a partial response was observed. A question arising out of this finding is whether administering the same dose more frequently would increase the response rate. The simulations have shown that minimal and maximal steady state concentrations of this dose given 2 times or 3 times a week were not much different. Therefore, the twice a week schedule for the future phase II study was selected, as it would be more convenient to patients.
| Confidential | - 60 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
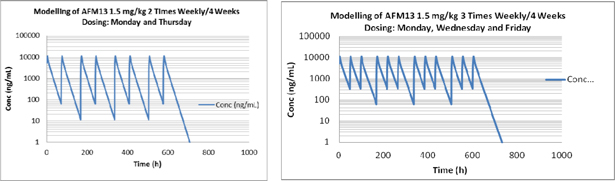
When comparing 4.5 mg/kg dosed twice a week and 3 mg/kg dosed 3 times a week, it appeared that 3 mg/kg dosed 3 times a week provided higher Cmin concentrations, whilst 4.5 mg/kg twice a week showed much higher peak, but lower trough concentrations. Assuming that the peak concentration on its own, as seen with 7 mg/kg, is not the main contributor to efficacy, 3 mg/kg 3 times a week for the phase II study were selected, as it provides the most consistent plasma concentrations.
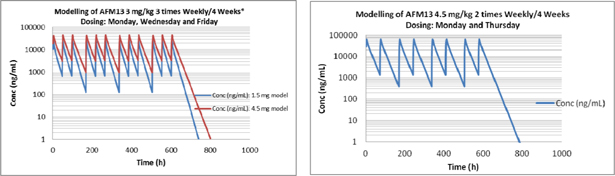
In conclusion, after various PK simulations the following two dose schedules will be used in the phase IIa study:
| 1. | 1.5mg/kg twice weekly, and |
| 2. | 3mg/kg 3 times weekly |
Based on these simulations, the differences between the anticipated minimal concentrations and maximal concentrations of the two regimens would be sufficiently wide to enable detection of the superior regimen.
Summary on the chosen Phase IIa study design and patient selection criteria
The proposed AFM13 Phase IIa study design is based on the Xxxxx design which uses a two stage design. This kind of clinical study design is widely used for the development of new drug candidates, especially in oncology.
In order to show the ³ 30% OR, Affimed will recruit during stage 1 of this Phase IIa 9 patients for each dosing schedule. If there is at least one OR in stage 1, stage 2 will be initiated, and additional 16 patients will be treated with that dose regimen of stage 1 which performed best. In total, 34 eligible patients are required to show the statistical significance for a ³ 30 %OR.
| Confidential | - 61 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
Based on very recent data of our AFM13 Phase I clinical trial we have to consider a ~15% “drop-out” rate. This means that Affimed will have to recruit up to 40 patients in order to achieve the lower limit of 34 eligible patients. The inclusion criteria for this Phase IIa will be more restrictive as compared to the Phase I, e.g. lower number of previous therapies (for further details please refer to section “patient criteria” above). However, all patients who will be included in this trial, had received as one of their prior therapies the antibody drug conjugate Adcetris.
Note: For more details and background information on the Phase IIa design based on Xxxxx, please refer to page 31ff.
VII) Conclusion on the further development of AFM13
Manufacturing. A robust and scalable manufacturing process is established which ensures the material supply for all clinical developmental steps as well as the early market needs. The Drug Product has a shelf life of more than ***** years, which is – for a biological therapeutic – remarkable.
Benign safety profile and encouraging efficacy in Phase I. Up to now (status March 2013) half of the patients treated with AFM13 showed clear signs of benefit. Moreover, in the 13 patients treated with 1.5mg/kg and higher doses, 2 partial responses and 7 stable diseases were observed. This encouraging data warrants the further development of AFM13 in phase II to assess its efficacy in better prognosis Hodgkin Lymphoma patients. In addition, the excellent safety profile of AFM13 is an especially attractive feature of this potential drug.
Medical need in HL. Especially the benign safety profile of AFM13 will allow a positioning in several therapeutic settings in HL. Although the cure rate of HL is relatively high, there is still a very high medical need for some patient groups: for elderly patients where chemotherapy is contra-indicated, for high risk patients with high relapse rate and finally for relapsed or refractory patients. At the moment no immunotherapeutic treatment is available for HL and the initially very impressive data of Brentuximab vedotin are – in the patients’ perspective – conflicting: very high response rates combined with severe side effects and a median duration of response of 6 to 7 months. Although targeted by an antibody, Brentuximab vedotin is “only” an additional chemotherapeutic with limited applicability in the treatment of HL patients. In this context, AFM13 has potential as 3rd line therapy for relapsed and refractory HL patients as well as 1st / 2nd line therapy. XXXx suggested that AFM13 should not only be developed in 3rd line as a salvage therapy but, in addition, in *****.
Development path and timelines. The starting point for the further clinical development is the production of GMP compliant AFM13 Drug Product and the recruitment for the Phase IIa study. However, since the GMP production process is established and since the recruitment of Phase I was faster than anticipated, that the timelines for the study would be: start in ***** and completion in *****.
| Confidential | - 62 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
Timelines & Milestones
*****
| Confidential | - 63 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
Budget (please provide a budget for each phase or stage of the project, replicate page as needed)
| Stage or Phase AFM13 | ||||||||
| Start Date: ***** | End Date: ***** | |||||||
| In US $ |
Justification | Funding Request |
Other Funding (internal or external) |
Total budget | ||||||||||
| FTE costs (Affimed) |
***** | ***** | ***** | |||||||||||
| CMC |
***** | ***** | ***** | |||||||||||
| Clin Phase II |
***** | ***** | ***** | |||||||||||
| Consulting |
***** | ***** | ***** | |||||||||||
| Total |
4,401,351 | ***** | ***** | |||||||||||
The costs comprise four categories (FTEs, CMC, clinical and others) totaling in *****. The funding request from LLS is ***** which corresponds to 3,236,288€/4,401,351$. The other ***** of the project costs are funded by Affimed. The material project activities are planned to be executed from *****.
Budget Justification
Project structure and process
The project can be segmented in the CMC process and the regulatory clinical process. Both processes are structured in a parallel fashion. As the two main activities overlap to a great extent, the budget is structured in only one phase. CMC process / production and all clinical activities are mainly performed by external service providers. Affimed has established a good and trustful relationship to its partners, so it can be assumed that the already selected and contracted service providers will be at Affimed’s disposal for the continuation of the AFM13 project. However, Affimed’s personnel will be responsible for initiating, monitoring and controlling all activities. Therefore the budget comprises
| Confidential | - 64 - |
| Therapy Acceleration Program Biotechnology Accelerator Project Proposal Version 1.4 |
|
*****
| Confidential | - 65 - |
COMPANY INFORMATION
Senior Management
Xxx Xxxxx, PhD; CEO
Xx Xxxxx joined Affimed in October 2010 as Chief Commercial Officer and since September 2011 leads the company as CEO. He has more than 18 years professional experience with an extensive background in general management, business development, product commercialization, fund raising and M&A. Prior to Affimed, Xx. Xxxxx was Chief Commercial Officer at Jerini AG and CEO of Jenowis AG. At Jerini AG he was responsible for BD and the market introduction of Firazyr. He also played a major role in the M&A process of Jerini AG with Shire. From 2000 – 2002 Xx. Xxxxx was General Manager and VP of the Molecular Medicine Division at Carl-Zeiss in Xxxx. Xx. Xxxxx began his professional career in 1993 at MorphoSys as scientist. In 1997 he became Director of BD and in 1999, VP of Licensing and BD.
Xx. Xxxxx received his PhD in chemistry and biochemistry from the University of Munich in 1991, and he also received an M.D. from the Technical University (TU) of Munich in 1997. From 1991 until 1993 Xx Xxxxx work as a post doc at the Dana-Faber Cancer Institute at the Harvard Medical School in Boston, USA.
Xxxxxx Xxxxxxxxx, PhD; CSO
Xx. Xxxxxxxxx joined Affimed in 2011 as Chief Scientific Officer. He has 20 years professional experience in the field of biotherapeutics research and development. Prior to Affimed, Xx. Xxxxxxxxx was a Senior Research Fellow at Boehringer Ingelheim Pharmaceuticals where he led antibody discovery efforts directed towards inflammatory and cardiovascular diseases. From 2002 to 2009 Xx. Xxxxxxxxx was at Xencor Inc. where he led translational research resulting in several therapeutic candidates targeting malignant and normal B cells. Prior to that he developed genomics technologies at Lynx Therapeutics and utilized phage display technology for the development of catalytic antibodies at Neurex Corporation.
Xx. Xxxxxxxxx received his PhD in biochemistry from Brandeis University for studies of GPCR structure-function relationships employing the visual pigment rhodopsin. Prior to this, he completed an MS degree in bioorganic chemistry at St. Petersburg’s State University, where he developed a multi-step synthesis of a birth-control drug lead. From 1991 until 1995 Xx. Xxxxxxxxx was a post doc at Genentech, Inc. engaged in quantitative studies of the contribution of secondary structural elements to global protein stability.
Xxxxxxx Xxxxxxx, PhD MBA;CFO
Xx. Xxxxxxx joined Affimed in 2005 as CFO. He has a strong track record as lead advisor in a variety of transactions and financings in the life sciences and health care sector.
Xx. Xxxxxxx is founder and CEO of MedVenture Partners — a Munich based corporate finance and strategy advisory company focusing on the life sciences and health care industry. Prior to founding MedVenture Partners, Xx. Xxxxxxx worked with KPMG for more than six years, where he was responsible for biotech and healthcare assignments. Before joining KPMG, he worked for Deutsche Bank AG. Xx. Xxxxxxx holds a graduate degree in business administration and a PhD in public health and is an active lecturer at the Odeon Center for Entrepreneurship of the Xxxxxx-Xxxxxxxxxx University, Munich.
| Confidential | 66 |
Xxxxxxxx Xxxxx, MD PhD; CMO
Xx. Xxxxx joined Affimed in 2006. He is a medical professional with 30 years of combined experience in clinical practice, medical science and drug development of biologics and small molecules.
Xx. Xxxxx has been responsible for almost 100 clinical trials (Phase I – IV). He acts as Managing Director of Pharma Integra Limited, a London-based Biotechnology consultancy, and was Chief Clinical Officer at Antisoma, where he was responsible for the design and implementation of the global program of clinical trials on anti-cancer drugs.
Prior to Antisoma, Xx. Xxxxx was European Head of Clinical Research and Development at the Japanese company Eisai. Before joining Eisai, he worked at Boehringer Ingelheim and held various academic and clinical positions.
Company Description
Company Profile
Affimed is a clinical stage biotech company developing unique TandAb® antibody therapeutics for cancer and inflammatory diseases. We have generated a comprehensive pipeline of antibody product candidates based on our proprietary TandAb® technology platform.
TandAbs are innovative bispecific tetravalent antibodies. This versatile technology produces biotherapeutic leads that possess drug-like properties with excellent product stability. A robust manufacturing process has been established. TandAbs promise increased therapeutic potential and superior safety profiles compared to monoclonal antibodies. Furthermore, when compared to antibody fragments and scaffolds, TandAbs show better targeting properties due their bivalent binding and have a much longer half-life.
Xxxxxxx’s lead candidates focus on highly attractive market opportunities. AFM13 proved its safety, tolerability and anti-tumor activity in a Phase I clinical trial in refractory or relapsed Hodgkin Lymphoma patients. AFM13 has a significant sales potential of *****. Another lead candidate is AFM11 for the treatment of Non-Hodgkin Lymphoma which is currently in late preclinical development. Further novel product candidates for the treatment of solid tumors and inflammatory diseases are in development.
Company History
Affimed was founded in 1999 as a spin-off from the German Cancer Research Centre (DKFZ) by Prof. Xx. Xxxxxx Xxxxxx in Heidelberg. Today, Affimed employs about thirty staff. Affimed has risen 65.5M€ so far and is backed by a peer group of investors including Orbimed, Aeris, LSP, BioMed Invest and Novo Nordisk A/S.
| Confidential | 67 |
Scientific Advisory Board
Safety Review Committee (SRC)
| • | Prof. Xx. Xxxxxxx Xxxxxx, Principal Investigator University Hospital of Cologne, Professor for Internal Medicine, Hematology & Oncology / Chairman, German Hodgkin Study Group, Germany |
| • | Prof. Xx. Xxx Xxxx, Principal Investigator University Hospital of Würzburg, Professor for Internal Medicine, Germany |
| • | Xx. Xxxxxxxx Xxxxx, Chief Medical Officer, Affimed Therapeutics AG, UK |
| • | Xx. Xxxxxxxxx Xxxxx, Consultant for Clinical Operations, syneed medidata GmbH, Germany |
| • | Xx. Xxxxxxx Xxxxxx, Medical Monitor, Drug Safety Specialist, spm2 Safety – Projects and more GmbH, Germany |
Data Monitoring Committee (DMC)
| • | Prof. Xx. Xxxxx Xxxxxxxxx, Chairman DMC, Professor for Hematology, University Medical Center Utrecht, Netherlands |
| • | Prof. Xx. Xxxxxx Xxxxxxxxxxxx, DMC Member, Professor of Hematology, CHRU de Lille, Hôpital Xxxxxx Xxxxxx, Service des Maladies du Sang, France |
| • | Prof. Xx. Xxxxxx Xxxx, DMC Member, Professor of Immunology and Immunotherapy, Director of Institute of Cellular Therapeutics, , Hannover, Germany |
Financial Information
The company was founded in 1999, it has risen 65.5M€ so far. The company is funded by a strong VC Syndicate: Aeris Capital, CH; BiomedInvest, CH; LSP, NL; NovoNordisk, DK; Orbimed, USA.
Affimed’s budget is sufficient to support the operations until the end of 2015 including the Phase IIa development of AFM13:
| • | A 15.5 M€ Series D financing was closed in September 2012. ***** |
| • | The budget includes an industry collaboration which leads to further revenues of 16.4 M€. |
| • | A 3.236 M€ / 4.401 M$ funding contribution by LLS |
Based on this budget the company is able to conduct the AFM13 clinical trial and finance the remaining budget, which would not be covered by the LLS funds.
| Confidential | 68 |
Company Intellectual Property
For the AFM13 project three patent families apply as listed below: The first patent “Multivalent antibody constructs” represents the TandAb core patent which protects all generated TandAbs. The “Anti-CD16A binding molecule” patent protects the effector mechanism, which specifically targets NK cells via binding exclusively to the isoform A of the CD16 receptor. The “CD16xCD30” patent is granted in Europe and protects the use of bispecific antibody constructs for the lysis of CD30 positive cells.
“Multivalent antibody constructs” (TandAb)
| • | Application discloses bivalent and tetravalent Fv antibody constructs (TandAb); constructs can be monospecific, bi- or multispecific. Each molecule comprises four variable domains linked by linkers 1, 2, and 3 which can differ in length; general diagnostic and therapeutic use, in particular for viral, bacterial or tumoral disease is mentioned |
| • | Patent term: May 5, 2019 |
| • | Granted in: Europe, USA, Japan, Australia, Canada |
“Anti-CD16A binding molecules”
| • | The invention relates to anti-CD16A binding molecules which are not binding to CD16B; various bispecific antibodies or different antibody formats and their medical uses are disclosed |
| • | Filing date: May 26, 2006 (patent term possible up to 2026) |
| • | Status: pending in Europe, USA, Australia, Canada, China, Russia, India, Brazil |
“CD16/CD30”
| • | Patent relates to bispecific CD16xCD30 Fv antibodies useful for the lysis of CD30 expressing cells (such as HL); exemplified are diabodies; not limited to particular format |
| • | Patent term: August 2, 2020 |
| • | Granted in: Europe |
AFM13 has FTO through Affimed’s IP and through licensing agreements with DKFZ (German Cancer Research Institute), Xoma (Phage Display for the generation of anti-CD30 and anti-CD16A antibodies which are incorporated in the AFM13 compound) and DadeBehring / Siemens (human antibody library generation).
| Confidential | 69 |
Competitive Intellectual Property Landscape
Introduction
Currently no other immunotherapeutic is available for the treatment of Hodgkin Lymphoma or other CD30+ positive malignancies. The recently marketed Seattle Genetics product Adcetris® (Brentuximab-vendotin) is labeled for use in refractory or relapsed Hodgkin Lymphoma patients. However, the IgG part of Brentuximab vedotin is only for targeting the drug; the cytotoxic activity is provided by an auristatine derivative. It is considered to be another chemotherapeutic approach. Several years ago, some real immunotherapeutic treatments were developed based on monoclonal antibodies, Fc-enhanced IgGs and bispecific antibodies, but these approaches were discontinued due to the lack of efficacy.
Antibody-drug-conjugates (ADCs)
Adcetris® (Brentuximab vedotin) is a CD30-directed antibody-drug conjugate indicated for (i) the treatment of patients with Hodgkin lymphoma after failure of autologous stem cell transplant (ASCT) or after failure of at least two prior multi-agent chemotherapy regimens in patients who are not ASCT candidates and (ii) the treatment of patients with systemic anaplastic large cell lymphoma after failure of at least one prior multi-agent chemotherapy regimen. These indications are based on objective response rate (XXX). There is no data available demonstrating improvement in patient-reported outcomes or survival with Brentuximab vedotin. XXX of Brentuximab vedotin in HL patients was 73% (CR: 32.5%; PR: 40.5%) with a median duration of response of 6.7 months. There is significant toxicity associated with Brentuximab vedotin of the following disorders: (i) blood and lymphatic system disorders (e.g. neutropenia), (ii) nervous system disorders (e.g. peripheral sensory neuropathy), (iii) general disorders and administration site conditions or (iv) gastrointestinal disorders. In addition, a significant proportion of patients showed grade 3 and 4 toxicities. In conclusion, although Brentuximab vedotin showed impressive XXX in salvage HL [and ALCL patients], its duration of response and its side effect profile are clear weaknesses that can be addressed by a safer and more durable therapy.
Monoclonal antibodies
BMS has investigated MDX-060, a fully human antibody targeting CD30. Preclinical studies suggest that MDX-060 antibody may induce tumor regression through direct inhibition of cellular proliferation or through the recruitment of immune effector cells. The chimeric antibody SGN-030 of Seattle Genetics has a similar mode-of-action. Recently, these antibodies showed disappointing results in phase II clinical studies with a small portion of partial remissions (0-6%) and stable disease. Both companies have therefore replaced these products with antibodies with improved effector function (MDX-1401) or with an anti-CD30-immunetoxin-conjugate (SGN-35). In addition, Xencor has developed an antibody with improved effector function (XmAb-2513). Neither the treatment with MDX-1401 nor XmAb-2513 lead to improved outcomes versus the predecessor antibodies and currently none of such antibodies is continued in further clinical studies.
Other bi-specific antibody constructs
Several years ago, two bi-specific antibodies were investigated in clinical trials by Biotest and Medarex, respectively. The natural killer cell-activating anti-CD16/CD30 bispecific monoclonal antibody (BiMAb) had shown efficacy in a Phase I/II trial of refractory Hodgkin Lymphoma. A total of 16 heavily pretreated patients received one to four BiMAb courses. Overall, 1 CR and 3 PR were observed. A bispecific molecule (H22xKi-4) comprising anti-CD30 and anti-CD64 F(ab’)
| Confidential | 70 |
fragments was investigated in patients with refractory HL. In the dose-escalation study ten patients were enrolled. Response to H22xKi-4 included 1 CR, 3 PR, and 4 SD. Both BiMab and H22xKi-4 appeared to be safe and well tolerated. Although both antibodies showed highly promising results in HL patients, the development of such compounds could not be continued due to manufacturing and scale-up issues.
So far, no immunotherapeutic has been approved for the treatment of Hodgkin Lymphoma. However, based on encouraging clinical results of a bi-specific (CD30/CD16) hybridoma antibody (Blood (1997) 89:2042 and Clin. Cancer Res. (2001) 7:1873), Affimed has developed a novel bispecific antibody, called AFM13.
Comparison of AFM13 with other immunotherapies
AFM13 has demonstrated superior ADCC properties on several Hodgkin Lymphoma and Anaplastic Large Cell Lymphoma (ALCL) cell lines relative to an Fc-enhanced anti-CD30 antibody. The EC50 has been increased by more than 10-fold. Moreover, in human serum the ADCC of AFM13 was similar to that in standard assay buffers, whereas the activity of the Fc-enhanced antibody was dramatically reduced; the reason is that the anti-CD16A moiety does not compete with polyclonal IgG in human blood and the binding of CD16A by TandAbs is not affected by Fcg receptor polymorphism, unlike existing therapeutic antibodies such as Rituximab. Therefore, we expect better clinical efficacy, lower doses with reduced treatment costs, and fewer side effects to achieve equivalent efficacy.
Comparison of AFM13 with Brentuximab vedotin
AFM13 has shown a better safety profile than Brentuximab vedotin. Although Brentuximab vedotin is based on an anti-CD30 antibody, its toxicity is primarily due to its chemical payload, the microtubule disrupting agent MMAE (monomethyl auristatin E). Brentuximab vedotin therefore shows a side effect profile similar to other chemotherapeutic agents. In contrast to this, AFM13 has shown a very benign safety profile in the dose-escalation trial with grade 1 and 2 infusion related side effects. AFM13 is further differentiating from Brentuximab vedotin due to its mode of action. While Brentuximab vedotin requires both CD30 internalization and actively dividing Xxxx Xxxxxxxxx cells, AFM13 simply relies on binding to CD30 positive cells and the engagement of NK-cells. Therefore, CD30 positive Xxxx Xxxxxxxxx cells can be killed without the requirement of CD30 internalization or active cell division. In particular, minimal residual diseases (MRD) can be addressed with AFM13. Due to the benign safety profile of AFM13, the drug should have a significant advantage over Brentuximab vedotin in a combination with existing chemotherapy in 1st or 2nd line therapy. In addition, AFM13’s different mode of action as compared to chemotherapy or Brentuximab vedotin can target non dividing malignant cells and minimal residual disease, which may contribute to time to relapse. ABVD and BEACOPP are established 1st and 2nd line therapies since many years; however, are also highly toxic. A combination of Brentuximab vedotin with ABVD could not be pursued due to pulmonary toxicity and Brentuximab vedotin is currently investigated in combination with AVD.
| Confidential | 71 |
Project Freedom-to-Operate Assurance
*****
Commercialization Plan
Following a successful outcome of the phase IIa study, AFM13 could be developed as a monotherapy to treat HL patients that have failed >2 lines of therapy or can be developed in *****.
| • | Development of AFM13 as monotherapy that have failed >2 lines of therapy |
Currently, only Brentuximab vedotin (Adcetris®) is registered for this indication and AFM13 may be given either prior to Brentuximab vedotin or after Brentuximab vedotin treatment. As Brentuximab vedotin has a short duration of response and cannot keep patients in remission long term or even cure patients, there is a high medical need for such patients.
| • | ***** |
*****
After completion of the phase IIa, Affimed plans to further develop AFM13 either on its own or in a partnership.
| • | Affimed would have to raise cash to fund the further clinical development. Our plans are to raise $50-70M through a public offering (IPO). We have tested the feasibility of such an offering by meeting with private and public investors. The feedback has been that, once positive phase IIa data are available, Affimed would be perceived as a candidate for an IPO. |
| • | Affimed recently met with >10 companies to discuss a partnership and there is a substantial interest in AFM13. However, at this stage the company prefers to perform the phase IIa on its own and partner at a later stage. Xxxxxxx’s goal is to enter a global partnership with a major player in the field of lymphomas either after phase IIa or prior to approval of the first indication. |
| Confidential | 72 |
ADDITIONAL INFORMATION
Citations
TandAbs and AFM13
XxXxxxxx et Eser: RECRUIT-TandAbs: harnessing the immune system to kill cancer cells. Future Oncol. 2012 Jun;8(6):687-95
Xxxxxxx et al: Rescue of Impaired NK Cell Activity in Hodgkin Lymphoma With Bispecific Antibodies In Vitro and in Patients. Mol Ther. 2013 Apr;21(4):895-903
Treatments in Hodgkin Lymphoma
Xxxx et Xxxxxx: Novel treatment strategies for patients with relapsed classical Hodgkin lymphoma. Blood Rev. 2010 Nov;24(6):233-8
Xxxxxx et al: Phase I/II Study of an Anti-CD30 Monoclonal Antibody (MDX-060) in Hodgkin’s Lymphoma and Anaplastic Large-Cell Lymphoma. Clin Oncol. 2007 Jul;25(1):2764-2769
Xxxxxxxxx et al: Phase 1 trial of the novel bispecific molecule H22xKi-4 in patients with refractory Hodgkin lymphoma. Blood. 2002 Nov;100(9):3101-3107
Xxxxxxxx et al: A phase 1 multi dose study of SGN-30 immunotherapy in patients with refractory or recurrent CD30_ hematologic malignancies. Blood. 2008 Feb;111(4):1848-54
Xxxxx et al: Safety and efficacy of brentuximab vedotin for Hodgkin lymphoma recurring after allogeneic stem cell transplant. Blood. 2012 Jul 19;120(3):560-8
Further clinical development
Xxxxx XX. The determination of the number of patients required in a preliminary and a follow-up trial of a new chemotherapeutic agent. J Chron Dis 1961; 13:346-353.
| Confidential | 73 |
Xxxxx Design in oncology studies
To determine the number of patients required for a phase II trial, different multistage designs have been developed. The first design was proposed by Xxxxx in 1961 and is still being widely used. It is a two-stage design which allows for the rapid rejection of an ineffective treatment at the end of the first stage, and provides an estimation of the success rate with a given precision, at the end of the second stage.
Xxxxx proposed a method to calculate the minimum sample size required to estimate the response rate with a given degree of precision, under the precondition that the study would be terminated early if there was 95% confidence that the response rate was less than a target response rate of interest (usually a response of 20%). In this classic design, 14 patients (corresponding to the response rate of 20%), or 9 patients (corresponding to the response rate of 30%) are treated initially, and if no responses are observed, the drug is considered inactive. On the other hand, if at least one response is observed, additional patients are enrolled and the response rate is estimated. Thus, the Xxxxx design combines elements of estimation and hypothesis testing.
In statistics, the hypothesis testing is generally organized around the concept of null and alternative hypotheses. Usually, the alternative hypothesis is what the investigator/sponsor hopes is true, and the null hypothesis is what the investigator/sponsor hopes is not true. It is essential that these two hypotheses are constructed in such a way that one or the other must be true. The data analysis focuses on testing the null hypothesis. If the null hypothesis is rejected (i.e., Ho: pr £ 5% versus, or Ha: pr³ 20%, or Ha: pr³ 30%), then one can accept the alternative hypothesis. Usually the null hypothesis states that there is no difference between two observations. Therefore, if this hypothesis is rejected, the alternative hypothesis that there is a difference between two observations is accepted. This approach has raised a lot of questions as the alternative hypothesis would be accepted even without being tested. Xxxxx and Xxxxxxx introduced different statistical approaches offering more outcome options. Nevertheless, even such designs were not sufficient to show that the alternative hypothesis is true in cases when the null hypothesis is rejected.
To overcome the problem of accepting the untested hypothesis, Xxxxx swapped the null and alternative hypotheses. So the hypothesis tested during the initial stage of the study is that the drug is active (the response rate is higher than a pre-determined minimal response rate of interest).This is the null hypothesis. If none out of 14 (for the difference of 20%), or 9 (for the difference of 30%) patients responds this hypothesis is rejected. Therefore, the alternative hypothesis, that the drug is not active enough to achieve a 20%, or 30% response, is accepted. In this design, the investigators/sponsor would hope that the null hypothesis is true, which is an unusual scenario for statistics in clinical trials.
| Confidential | 74 |
It is accepted that two types of error can occur when applying statistics in clinical trials:
| • | Type I error, an a error, or a “false positive”, is the error of rejecting a null hypothesis when it is actually true. In order to reduce this type of error, the convention is to take a small a value (i.e. 5%). |
| • | Type II error, a b error, or a “false negative” is the error of failing to reject a null hypothesis when it is in fact not true. In this type of error the b value is accepted as being 10%. |
Affimed’s calculation follows these principles and for Stage 1 Affimed suggests selecting the sample size based on the precision (a) of 5%. For the phase IIa study, the sample size for Stage 2, it is suggested adopting the lower precision of 10%. However, for the phase IIb it should be considered to adopt again the higher precision of 5%.
Two Stage Xxxxx Design:
| • | No control (only historical data) |
| • | Two stages (double sampling) |
| • | Goal is to reject ineffective drugs ASAP |
| Decision I: | Drug is unlikely to be effective | in ³ x% of patients | ||
| Decision II: | Drug could be effective | in ³ x% of patients |
| A. | Let x% = 20% (drug likely to work in at least 20% of patients) |
| 1. | Enter 14 patients |
| 2. | If 0/14 responses, stop and declare true drug response £20% |
| 3. | If ³1/14 responses, add 15-40 more patients |
| 4. | Estimate response rate & 95% C.I. |
| B. | Let x% = 30% (drug likely to work in at least 30% of patients) |
| 1. | Enter 9 patients |
| 2. | If 0/9 responses, stop and declare true drug response £30% |
| 3. | If ³1/9 responses, add 70-91 (for SE5%), or 11-16 (for SE10%) more patients |
| 4. | Estimate response rate & 95% C.I. |
Stage 1 Sample Size
| Rejection Error |
Effectiveness (%) | |||||||||||||||||||||||||||
| 5 | 10 | 15 | 20 | 25 | 40 | 50 | ||||||||||||||||||||||
| 5% |
59 | 29 | 19 | 14 | 11 | 6 | 5 | |||||||||||||||||||||
| 10% |
45 | 22 | 15 | 11 | 9 | 5 | 4 | |||||||||||||||||||||
Stage 2 Sample Size
Based on desired precision of effectiveness estimate
r1 = # of successes in Stage 1
n1= # of patients in Stage 1
| Confidential | 75 |
Additional Patients for Stage 2 (n2)(Rejection Rate 5% for Stage 1)
| Required Precision (SE) |
Number of Successes Stage I |
Therapeutic Effectiveness (%) |
||||||||||||||||||||||||
| 5 | 10 | 15 | 20 | 25 | 30 | |||||||||||||||||||||
| ±1 SE 5% |
n1 | 59 | 29 | 19 | 14 | 11 | 9 | |||||||||||||||||||
| 1 | 0 | 4 | 30 | 45 | 60 | 70 | ||||||||||||||||||||
| 2 | 0 | 17 | 45 | 63 | 78 | 87 | ||||||||||||||||||||
| 3 | 0 | 28 | 58 | 76 | 87 | 91 | ||||||||||||||||||||
| 4 | 0 | 38 | 67 | 83 | 89 | 91 | ||||||||||||||||||||
| 5 | 0 | 46 | 75 | 86 | 89 | 91 | ||||||||||||||||||||
| ±1 SE 10% |
1 | 0 | 0 | 0 | 1 | 7 | 11 | |||||||||||||||||||
| 2 | 0 | 0 | 0 | 6 | 12 | 15 | ||||||||||||||||||||
| 3 | 0 | 0 | 1 | 9 | 14 | 16 | ||||||||||||||||||||
| 4 | 0 | 0 | 3 | 11 | 14 | 16 | ||||||||||||||||||||
| 5 | 0 | 0 | 5 | 11 | 14 | 16 | ||||||||||||||||||||
*****
| Confidential | 76 |
EXHIBIT E
Report Schedule
| DELIVERABLE |
DUE DATE | |
| Progress report of the AFM13 Development Program since the prior AFM13 RAC meeting, as described in Section 2.7(a). | Within ***** prior to each AFM13 RAC meeting | |
| Financial reports, as described in Section 2.7(b). | Within ***** after the end of each fiscal year ending prior to the Program Termination Date and within ***** after the fiscal quarter in which the Program Termination Date occurs. | |
| A Final Progress Report, as described in Section 2.7(c). | Within ***** after the Program Termination Date. | |
| Notice of any license, sublicense or transfer of any Program Invention or Development Program Results, or subcontract or permitted assignment by Affimed of this Agreement or its rights and/or obligations hereunder, or of any Change of Control Transaction, as described in Section 2.7(d). | ***** during the AFM13 Development Program and thereafter. | |
| Notice of all material actions, suits, claims, proceedings, investigations and inquiries that directly or indirectly involve or impact Affimed, as described in Section 2.7(e). | ***** | |
| Progress reports and status updates on Affimed’s activities with respect to the Product, as described in Section 2.7(f). | *****, and in any event within ***** after January 1 and June 1 of each fiscal year following the Program Termination Date until First Commercial Sale. |
| Confidential | 77 |

