
PROJECT ADDENDUM 1 TO MASTER SERVICES AGREEMENT
Exhibit 10.13

PROJECT ADDENDUM 1 TO MASTER SERVICES AGREEMENT
This Project Addendum is effectively dated as of October 9, 2019 (the “Effective Date”) by and between Blue Water Vaccines, Inc. a Delaware corporation located at ▇▇ ▇▇▇▇ ▇▇▇▇▇▇ ▇▇▇▇▇▇, ▇▇▇▇▇ ▇▇▇, ▇▇▇▇▇▇▇▇▇, ▇▇ ▇▇▇▇▇ (“Blue Water”) and Ology Bioservices, Inc., a Delaware corporation having a principal place of business at ▇▇▇▇▇ ▇.▇. ▇▇▇▇ ▇▇▇▇▇, ▇▇▇▇▇▇▇, ▇▇▇▇▇▇▇ ▇▇▇▇▇ (“Ology Bio”). Blue Water and Ology Bio are sometimes referred to herein individually as a “Party” and collectively as the “Parties.”
WHEREAS, Blue Water and Ology Bio entered into a Master Services Agreement effectively dated as of July 19, 2019, (the “MSA”) whereby Ology Bio agreed to provide from time to time services and deliverables associated therewith (“Services”) to Blue Water pursuant to the terms and conditions set forth in the MSA and any Project Addendum;
WHEREAS, Blue Water and Ology Bio desire to enter into this Project Addendum for Services as set forth herein, subject to and in accordance with the MSA.
WHEREAS, Ology Bio submitted a Proposal dated May 22, 2019 to Blue Water.
NOW, THEREFORE, in consideration of the foregoing and the mutual promises, covenants and agreements set forth below, and for other good and valuable consideration, the receipt and sufficiency of which the parties hereby acknowledge, the parties agree as follows:
1.0 SCOPE OF WORK
Ology Bio will perform Process Development and CGMP Manufacturing of Recombinant Influenza Conserved Regions for Vaccine Production for Blue Water Vaccines, Inc. (Blue Water). Blue Water is currently developing a novel influenza vaccine based on conserved regions of the influenza virus. Each of these conserved regions encodes a unique 15-20 amino acid peptide sequence. Blue Water has demonstrated the each of these peptides is highly immunogenic and conserved over many different strains of influenza. Currently there are six (6) different conserved regions that are being developed. These peptides are currently being combined into two separate expression vectors in E.coli. Each of the two different combinations represent a single drug product. One will be used as the prime vaccine dose and the second will be used as the boost dose. The present scope of work includes the timely process development and manufacture of the two novel influenza vaccine drug substance candidates and production of the two different drug product candidates suitable for pre-clinical animal studies and Phase 1 clinical studies.
This project consists of eight Tasks required to manufacture and release a CGMP lot of Drug Substance (DS) and Drug Product (DP) suitable for Clinical Development and conduct preclinical IND-enabling studies and prepare the IND. Specific details of each task are provided in Section 2.0.
Task 1: Technology Transfer and Process Establishment
Task 2: Analytical Assay Development
Task 3: CGMP Master and Working Cell Banking
Task 4: Process Development and Scale-up
Task 5: Engineering Run and Stability Testing
Task 6: CGMP Run and Stability Testing
Task 7: Engineering Drug Product and Stability Testing
Task 8: CGMP Drug Product and Stability Testing
Task 9: Regulatory Support for Preclinical IND-Enabling Studies and IND Preparation
Page 1 of 22

2.0 TECHNICAL APPROACH AND PLAN
Task 1: Technology Transfer and Process Establishment
Table 1. Task 1 Technical Assumptions
| Technical Assumption(s) |
| Product-specific information (e.g., ▇▇▇▇ of Materials, previous run data, intermediate stability data, any regulatory documentation, test methods) will be transferred within 5 business days of contract signing. |
| The kick-off meeting will be scheduled within 10 business days of contract signing. |
| Two Process Establishment runs will be performed at this stage using the established methodology provided by Blue Water. |
| The upstream and downstream processing is similar between the different peptide constructs |
| Blue Water will provide Research Cell Bank vials, reagents and standards and associated Certificates of Analysis, as |
| required, to Ology Bio within 5 days of contract signing |
Information Transfer and Gap Assessment:
The Information Transfer stage is critical for the success and timeliness of the project. Ology Bio requests that all pertinent documents from Blue Water will be supplied within 5 days of contract signing to allow enough time for critical review by the Ology Bio team. A kick-off meeting will be scheduled with review of the plans and timelines. After the project gap analysis is complete, a final schedule and ▇▇▇▇▇ chart will be completed.
Receiving Blue Water Documentation:
To initiate the Technology Transfer, we will conduct a thorough review of all process and analytical documents provided by Blue Water. At this time, it will be agreed upon by Blue Water and Ology Bio that the manufacturing processes are similar enough between the different peptides that process development for only one will be sufficient and can be transferred to the other drug substance candidate. In collaboration with Blue Water, we will create a Development Plan for each of the drug substances and governance process to meet the objectives of the project. Blue Water will provide Ology Bio with all applicable standard operating procedures (SOPs), process procedures, process transfer protocols, analytical plans, specifications and other knowledge to transfer analytical methods and the manufacturing process. Technology Transfer will include the following preparation activities:
| ■ | Preparation of a Development Plan |
| ■ | Preparation of documentation |
| ■ | Equipment Identification |
| ■ | Flow diagrams as appropriate |
| ■ | Process step descriptions |
| ■ | Risk Analysis and Mitigation Strategy |
Page 2 of 22
Transfer of Product-Specific Materials from Blue Water and Procurement of Materials and Components:
Blue Water will provide Research Cell Bank (RCB) vials, reagents and standards, and associated Certificates of Analysis (COAs), as required, to Ology Bio within 5 days of contract signing in order to stay within the aggressive timeline for this program.
A full list of raw materials will be developed and sent to Blue Water for approval. All consumables, expendables and raw materials will be purchased using QA-approved vendors, properly inventoried and stored in the proper conditions. We may elect to purchase pre-prepared media and certain buffers from agreed-upon suppliers to avoid any variability in these critical reagents in the process. We will identify and qualify= suppliers of production materials and any required excipients. The nature of this project will require that additional materials identified in the Process Development Task to be communicated to Blue Water at a later date.
Development Plan and Reports:
Weekly or biweekly presentations will be provided to Blue Water that summarizes the performance of the process per plan. At the end of Task 1, a draft Development Plan will be written and reviewed/approved by both Ology Bio and Blue Water. The Development Reports will be written after execution of Establishment Runs to contain details on each unit operation; trending of the data compared with any available historical data (i.e., provided in Blue Water documents); analytical testing results; process deviations and impact assessment; our proposed process changes, including assessment of impact and justifications for changes; and updated risk and gap assessment.
Process Establishment:
Ology Bio will perform two Process Establishment Runs (one for each of the two candidates) to demonstrate the process for the Blue Water influenza vaccine candidates at 3 L fermenter scale and prepare a process transfer final report for approval by Blue Water. Ology Bio will use the existing RCBs from Blue Water to perform these runs. Upstream and Downstream Process Flow Diagrams for the production of the Blue Water influenza vaccine candidate are shown in Figure 1 and Figure 2.
Limited testing will be performed on these Establishment runs. These will include SDS- PAGE, protein concentration and HPLC analysis for purity. We will also present a preliminary ▇▇▇▇ of Materials (BOM) at the end of this task.
A list of deliverables for Task 1 is shown in Table 2.
|
Figure 1. Upstream
Fermentation
Figure 2. Downstream Process Flow Diagram 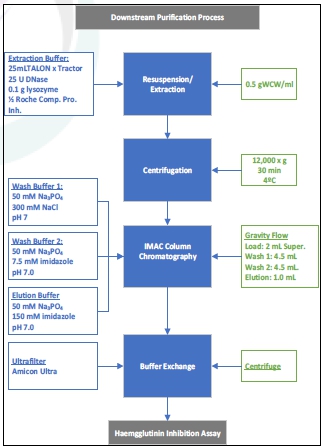 |

Page 3 of 22
Table 2. Task 1 Deliverables
| Task | Deliverable |
| Task 1: Technology Transfer and Process Establishment | Final Schedule Agreement with Blue Water and Ology Bio at completion of this stage |
| Project Management Plan including Project Charter | |
| Meeting agenda and minutes | |
| Process development plan | |
| Process establishment plan and report | |
| Preliminary ▇▇▇▇ of Materials |
Task 2: Analytical Assay Development
Table 3. Task 2 Technical Assumptions
Technical Assumption(s) |
| Blue Water will provide analytical Reference Standards, product-specific reagents (antibodies) and initial samples of DS and DP for use in method transfer and validation |
| All required analytical methods are summarized in Table 4 |
| Assay qualification will be phase appropriate |
| Forced degradation studies will not be required on DS |
A complete list of analytical assays will be provided by Blue Water or agreed upon with Ology Bio. Specifications for each of the assays will also be provided by Blue Water or agreed upon with Ology Bio. We propose to perform Technology Transfer feasibility assessments on the QC assays outlined in Error! Reference source not found. for in-process (IP) testing and DS testing. Each of these assays will need to be established for each of the two different drug substance candidates. Stability and DP testing are described in Tasks 6 and 7, respectively. Following the Technology Transfer feasibility assessment, QC scientists will revise method SOPs (as required) and verify methods for testing. Upon completion of the verification studies, a comprehensive report, reviewed and approved by QA, will be provided documenting the results of the verification, the suitability of the intended method, a description of test samples, a description of experiments and a summary of data for each parameter tested, as well as relevant raw data obtained from these studies. These assays will also be qualified as suitable for CGMP release and use in Phase 1 clinical trials. Testing will not be outsourced without the prior written consent of Blue Water.
Table 4. In-Process and Drug Substance Release Testing
| Assay | Method | Location | Specification | Process Step |
| Physiochemical Properties | ||||
| Appearance |
Visual Observation |
Ology Bio | Clear, colorless liquid; no particles |
DS |
| pH | USP<791> | Ology Bio | TBD | IP, DS |
| Conductivity | TBD | Ology Bio | TBD | IP |
| Viability | Cell count | Ology Bio | TBD | IP |
| Safety | ||||
| Endotoxin | USP<85> | Ology Bio | < 10 EU/dose; dose = 100 µg |
IP, DS |
| Bioburden | Membrane Filtration |
Ology Bio | < 10 CFU/mL | IP, DS |
| General Safety | 21 CFR 610.11 | Ology Bio | Pass | DS |
| Content | ||||
| Protein Concentration | BCA | Ology Bio | Report | IP, DS |
Page 4 of 22
| Assay | Method | Location | Specification | Process Step |
| Identity | ||||
| Presence of Blue Water Vaccine Candidate1 | Western | Ology Bio | Identity Confirmed | IP, DS |
| Purity | ||||
| Purity | SDS-PAGE | Ology Bio | TBD | IP, DS |
| Host Cell DNA | E. coli qPCR assay |
Ology Bio | TBD | DS |
| Host Cell Protein | Kit | Ology Bio | TBD | DS |
| Potency | ||||
| In vitro immunopotency | ELISA | Blue Water | TBD | DS |
1Assay will be Tech Transferred to Ology Bio
A list of deliverables for Task 2 is shown in Table 5.
Table 5. Task 2 Deliverables
| Task | Deliverable |
| Task 2: Analytical Assay Development | Analytical assay Qualification Plan for each analytical method for each of the two different drug substance candidates |
| QA-reviewed and approved Qualification Report for each analytical method | |
| In-process and release assay specifications |
Task 3: CGMP Master and Working Cell Banking
Table 6. Task 3 Technical Assumptions
| Deliverable |
| A minimum of 300 CGMP MCB vials will be prepared for each of the two different drug substance candidates in support of this project |
| A minimum of 300 CGMP WCB vials will be prepared for each of the two different drug substance candidates in support of this project |
| RCB required for MCB production will be generated at Ology Bio in Task 4 |
In compliance with CGMP Regulations, Ology Bio will produce a minimum of 300 vials of a Master Cell Bank (MCB) for each of the two different drug substance candidates per QA-approved batch production records using the RCBs generated by Ology Bio as part of Task 4. The new Blue Horizon vaccine candidates will not contain a His-tag. These RCBs will be utilized for the production of the CGMP MCBs. The new MCBs will undergo characterization and release testing based on an analytical control strategy outlined in Table 7. Ology Bio proposes to generate CGMP Working Cell Banks (WCBs) from the MCBs of each of the two different drug substance candidates and characterize them.
Page 5 of 22
Table 7. Cell Bank Release Assays
| Cell Bank Release Assay |
| Strain ID/Purity-Differential Media |
| Product ID-Dot or Western or SDS-PAGE |
| Viability |
| Growth Stability |
| Plasmid Sequencing |
| Plasmid Size |
| Genetic Stability-Copy Number |
| Genetic Stability-Plasmid Restriction Analysis |
| Plasmid Retention/Antibiotic Resistance |
| Bacteriophage |
An annual stability program will also be initiated and will be continued for up to three years for both MCB and WCB. For budgeting purposes, testing will only be conducted for 12 months, but cells will remain on stability for future testing. The stability testing is provided in Table 8.
Table 8. Cell Bank Stability Assays
| Cell Bank Stability Assays |
| Growth Stability |
| Viability |
| Genetic Stability-Plasmid Restriction Analysis |
| Plasmid Retention/Antibiotic Resistance |
Table 9. Task 3 Deliverables
| Task | Deliverable |
| Task 3: CGMP Master and Working Cell Banking | Summary Report on cell banking and release for both MCB and WCB for each of the two drug substance candidates including COAs and copies of Master Batch Records |
| Cell bank stability plan for each of the two candidate MCB and WCB for review and approval by Blue Water | |
| A minimum of 300 CGMP MCB vials for each of the two drug substance candidates | |
| A minimum of 300 CGMP WCB vials for each of the two drug substance candidates |
Task 4: Process Scale-up and Optimization
Table 10. Task 4 Technical Assumptions
| Technical Assumption(s) |
| The Process Development Plan outlining the experimental plan will be approved by Blue Water |
| Successful Process Establishment Runs were completed in Task 1 |
| Process Development will include development of non-His-tagged influenza vaccine candidates |
| Process Development will include generation of new RCBs for each of the two drug substance candidates |
| Process Development will be performed on both drug substance candidates |
| Six process development runs (three for each candidate) at the 3 L scale will be performed |
| Two scale-up runs at the 120 L scale will be performed |
Page 6 of 22
The new plasmids provided by Blue Water will be evaluated using two different E. coli expression systems: one based on IPTG induction, the other based on phosphate depletion (PhoA). The expression system that yields the highest soluble titer will be selected for further development. We will initially generate RCBs. We will identify three individual colonies from each of the two different drug substance candidates, and each of the colonies will be analyzed for protein production at the 1-3 mL scale. Two colonies will be scaled up and used to generate 50 vials of RCB for each candidate. These RCBs will then be utilized for production of the CGMP MCBs and WCBs as described in Task 3. Each RCB will be characterized as per Table 11.
Table 11. RCB Release Assays
| Release Assays |
| Viability |
| Growth Stability |
| Plasmid Size |
| Genetic Stability-Plasmid Restriction Analysis |
These RCBs will be used for the initial process development studies. The process development will include both candidates and on both the upstream and downstream processing of the vaccine candidates. Small-scale upstream production runs (1 L scale) will be used to generate materials for the downstream processing. Ology Bio proposes to use their rapid chromatography screening protocols to identify and optimize the chromatographic procedures required for the purification of the Blue Water vaccine candidate. Ology Bio proposes to scale-up the manufacturing process to the 120 L scale. This will include six 3 L production runs (three for each candidate) to identify the optimum upstream parameters for maximum production of the vaccine candidate. In parallel, the downstream processing will be optimized as described above and then scaled to the 3 L scale. Ology Bio then proposes to perform two scale-up runs at the 120 L scale using the 150 L stainless steel fermenter, the proposed manufacturing production scale. These runs will be analyzed using the in-process and release tests as described in Task 2. Success criteria for these runs will be agreed upon by Blue Water and Ology Bio prior to initiation of these runs. The sampling plan for these runs will also be agreed upon prior to initiation of the runs. Draft batch records and a final sampling plan will be prepared for use in the Engineering runs (Task 5).
Equipment required to support all the required tasks for the program are in-place and have been previously qualified at the ADM Facility, as listed in Table 12. Should additional equipment be required, qualification will be performed per standard operating procedures prior to CGMP manufacturing.
Table 12. Equipment list for DS Manufacturing at the ADM Facility
| Item | ADM Equipment Identification |
| Analytical Scale | ▇▇▇▇▇▇ XS2002S |
| Autoclave | Fedegari Autoclave Model NA2420AW |
| Centrifuge | Sorvall LYNX 6000 |
| Freezer | Revco High Performance -20° C Freezer |
| Mixing Vessels | GE XDM-Quad Mixing Systems (50-500 L) |
| Depth Filtration Systems | Millipore POD, 3M and Sartorius Holders available |
| Chromatography System | GE Akta Ready |
| TFF System | Sartorius Sartoflow Alpha Plus and Flex Act |
| Fermenter | Eppendorf BioFlo Pro 120 L and 60 L |
| Incubator Shaker | Eppendorf I42R |
| Mini Fermenter System | Eppendorf |
| Microscope | ▇▇▇▇▇▇ LMI-6001B |
| Pump(s) | W/M Peristaltic Pump, 520SN/R2; |
Page 7 of 22
| Item | ADM Equipment Identification |
| W/M Peristaltic Pump,520 Um AN/R2; W/M Peristaltic Pump, 323S/RL2 | |
| Refrigerator | REL4504A Revco High Performance Refrigerator |
| Scale | ABSCO SIWSDCS-1-35H-E7 |
| Water Bath(s) | ▇▇▇▇▇▇ Scientific Model 265/2866 |
| Vapor Phase Liquid Nitrogen (LN2) Freezer | Forma 7402 CryoPlus |
| Stir Plate | Corning PC-240 |
| pH meter | Orion Versa Star Meter, pH/Conductivity |
| Balance | ABSCO SIWSDCS-1-35H-E7 |
| Conductivity Meter | Orion Versa Star Meter, pH/Conductivity |
| Class B Laminar Flow | NuAire BSC NU-430-600 |
| Pipette Aid | Numerous |
| Micropipettor (200μL) | Numerous |
| Micropipettor (1000μL) | Numerous |
| Freezer ≤ -70° C | Thermo UFX600 |
| Autoclave for ▇▇▇▇▇▇▇▇▇▇▇▇▇▇▇ | ▇▇▇▇▇▇▇▇ ▇▇▇▇▇▇▇▇ |
| Multipette | Numerous |
| Balance | ABSCO SIWSDCS-1-35H-E7 |
Deliverables for this task are provided in Table 13.
Table 13. Task 4 Deliverables
| Task | Deliverable |
| Task 4: Process Development and Scale-Up | Process Development Report for Blue Water review and approval |
| Material from the Process Development Runs | |
| Process scale-up final report for review and approval by Blue Water | |
| Materials from the Scale-up runs | |
| Process parameters for the Engineering run | |
| Technology Transfer Protocol | |
| Draft batch records and sampling plan for use in the Engineering run | |
| Confirmation Run report and materials |
Task 5: Engineering Run and Stability Testing
Table 14. Task 5 Technical Assumptions
| Technical Assumption(s) |
| Ology Bio proposes to perform one Engineering run for each of the two drug substance candidates (total of 2 runs) at the 120 L scale |
| In-process and release testing will be performed as described in Task 3 |
| Materials will be placed on 12-month stability |
| DS Reference Standard materials will be generated (1,000 vials) for each of the six drug substance candidates |
We will perform one Engineering run for each of the two drug substance candidates at full scale of 120 L (total of two engineering runs). The in-process and release testing plan for this run will be agreed upon by Blue Water and Ology Bio. The Engineering run will be executed at the 120 L production scale by manufacturing staff in the CGMP manufacturing core at the Facility. The Engineering run will be performed using draft Master Batch Records and QA-released raw materials and components. The batch records will be redlined during the Engineering run, and any changes will be incorporated into the Master Batch Records prior to approval for CGMP manufacturing. The in-process and release assays are described in Task 3, Table 4.
Page 8 of 22
The material generated from this lot will be indicative of the CGMP-manufactured material. The materials from this lot will be made available to Blue Water for additional studies. In addition, materials from this lot will be used to generate Reference Standard materials. A COA and Material Safety Data Sheet (MSDS) will be prepared at this stage. A completed BOM will be submitted as part of this Stage. The non-CGMP DS material will be placed on stability studies.
Stability Testing:
Stability testing of non-CGMP Engineering DS will be conducted in accordance with current U.S. FDA Code of Federal Regulations (CFR) and International Conference on Harmonization (ICH) guidelines, including:
| ■ | 21 CFR Parts 210 and 211 (CGMPs) |
| ■ | 21 CFR Part 312 (IND Application) |
| ■ | ICH Q1A (R2) Guideline: “Stability Testing of New Drug Substances and Products,” February 2003 |
| ■ | ICH Q1C Guideline: “Stability Testing of New Dosage Forms," November 1996 |
| ■ | ICH Q5C Guideline: “Stability Testing of Biotechnological/Biological Products,” November 1995 |
The stability evaluation will support the following:
| ■ | Use of the investigational product throughout nonclinical studies and clinical trials |
| ■ | Mitigation of shipping and storage temperature excursion impact on the investigational product |
| ■ | Stability of the product during handling (clinical sites, emergency-use scenarios) |
| ■ | Selection of lot release and stability-indicating analytical test methods |
| ■ | Expiration or retest dates for DS |
| ■ | Product conformity to stability specifications throughout the clinical trial |
The proposed stability study for non-CGMP (as well as CGMP) DS lots is provided in Table 15. Stability testing will be conducted per approved protocols and reported annually.
Table 15. Drug Substance Stability
| Test | Location | Acceptance Criteria | 0m | 1m | 3m | 6m | 9m | 12m |
| pH | Ology Bio | TBD | X | X | X | X | X | X |
| BCA | Ology Bio | TBD | X | X | X | X | X | X |
| SDS-PAGE/Western blot/ densitometry to monitor protein degradation | Ology Bio | TBD | X | X | X | X | X | X |
| Sterility | Ology Bio | No fungal or bacterial growth observed | X | X |
Page 9 of 22
Reference Standard:
Providing high-quality, documented and qualified Reference Standards is critical to every batch released, and characterization can be an arduous process. In accordance with our QS, Reference Standards are produced and qualified prior to use in lot release, characterization or stability testing. The objective of this program is to provide complete documentation of the establishment and trending of product Reference Standards. Another goal of the program is to assess the suitability and availability of Reference Standards and critical reagents to meet ICH and FDA guidelines appropriate for the product lifecycle stage for which the materials will be used. This program results in complete documentation of these Reference Standards by providing:
| ■ | Manufacture according to approved batch record or protocol |
| ■ | Qualification according to approved qualification protocol |
| ■ | Lot release testing according to approved technical specification |
| ■ | Generation of a COA detailing the lot release testing results |
| ■ | Controlled storage conditions and inventory |
| ■ | Stability testing |
| ■ | Continual data trending and evaluation of suitability in new/revised analytical methods and/or with changes to manufacturing process operations |
For the Blue Water program, interim DS Reference Standards will be established in accordance with an approved protocol from the DS Engineering lot(s). To reiterate the approach to DS Reference Standard, a minimum of 1,000 vials of Engineering DS will be aliquoted and qualified as the DS Reference Standard. The filled Engineering DP generated in Task 7 will be labelled and qualified as the DP Reference Standard. A QA-reviewed and approved Qualification Report will be provided that documents suitability of the DS and DP Reference Standards in the intended methods. These interim DS and DP Reference Standards are to be used for Phase 1/2 product lot release and stability testing.
A list of deliverables for Task 5 is shown in Table 16.
| Table 16. Task 5 Deliverables | |
| Task | Deliverable |
| DS Engineering Report | |
| DS Engineering Stability Protocol/Report | |
| Finalized BOM | |
| Task 5: Engineering Run and | Finalized CGMP batch record templates |
| Stability Testing | Finalized CGMP DS Specifications |
| Engineering non-CGMP DS COA and MSDS | |
| Updated Tech Transfer Protocol (if needed) | |
| Engineering non-CGMP DS Products (two total) | |
Task 6: OPTIONAL – CGMP Run and Stability Testing
Table 17. Task 6 Technical Assumptions
| Technical Assumption(s) |
| Ology Bio will use the CGMP WCB |
| Ology Bio will perform one CGMP DS lot for each of the two drug substance candidates (total of two runs) at the 120 L scale |
Blue Water and Ology Bio will agree on the analytical and IP testing plan for the routine production of the Blue Water vaccine candidate, which we will implement. The manufacturing processes that were developed and generated in Task 5 will be used for both drug substance candidates. Following completion of the scalability studies, a Technology Transfer Protocol will be generated. This report will describe the process development and define the critical process parameters and established ranges. The report will summarize lot testing and establish a sampling and testing plan to be used during CGMP manufacturing. A BOM listing all required raw materials and components will be included. From this BOM, specifications will be created for each material, as well as for IP intermediates where required. Batch records will be finalized and approved by QA for use in the CGMP manufacturing campaign. Any changes identified during the execution of the DP Engineering run (Task 7) will be incorporated into the final CGMP batch records prior to execution.
Page 10 of 22
We will perform one CGMP manufacturing campaign in accordance with:
| ■ | 21 CFR Parts 210 and 211 (CGMP) | |
| ■ | 21 CFR Part 312 (IND Application) | |
| ■ | 21 CFR Parts 600 and 610 (Biological Products) | |
| ■ | 21 CFR Part 11 (Electronic Records and Signatures) |
To lead to a successful campaign, we will use manufacturing readiness reviews to ensure that all activities are completed prior to the start of manufacturing. We perform Area Clearance and Product Changeover according to internal SOP-09-00054. Trained Operations personnel will clear manufacturing areas after manufacturing campaigns following work instruction WI-09-00004. Areas are cleaned according to SOP-09-00006, including chlorine dioxide decontamination when appropriate. Activities are documented on forms and manufacturing areas are released for use after QA review of these activities.
Our QA takes responsibility for assuring the quality and integrity of products and all data generated in compliance with the FDA GLPs and CGMPs. QA provides review of manufacturing and testing operations as well as approval of specifications, Master Batch Records, procedures, contract manufacturers, system and equipment changes, and intermediate and final product release. Deviations and investigations are integrated in a corrective action system. QA review and approval activities will be carried out in support of CGMP production campaigns on all the following activities:
| ■ | Documentation: | |
| ◌ | Raw Material Specifications |
| ◌ | Manufacturing Master and Executed Batch Records |
| ◌ | Equipment Operation and Maintenance SOPs |
| ◌ | Analytical Method SOPs |
| ◌ | Cleaning and Disinfection SOPs |
| ◌ | Harvest and Final Product Specification |
| ■ | Manufacturing Cleanroom Preparation: | |
| ◌ | Cleaning of Laboratory during Manufacturing |
| ◌ | Area Clearance and Product Changeover |
| ◌ | QA Audits |
| ◌ | Cleaning Validation Risk Assessment/Protocol, if applicable |
| ■ | Reference Standard: | |
| ◌ | Certification of Reference Standard |
| ■ | Raw Materials: | |
| ◌ | Vendor Qualification |
| ◌ | Ordering of Raw Materials and Supplies |
| ◌ | Sampling of Raw Materials |
| ◌ | QC Testing of Raw Materials and Data Review |
| ◌ | QA Audit of Testing Data and Raw Material COAs |
| ◌ | QA Release of Raw Materials and Inventory Tracking |
| ■ | CGMP Manufacturing: | |
| ◌ | Manufacturing of DS | |
| ■ | Product Testing: |
Page 11 of 22
| ◌ | QC Review of Testing Data |
| ◌ | QA Review of Testing Data and Preparation of COAs |
| ◌ | QA Release of DS Lot(s) |
| ■ | Packaging and Shipping: | |
| ◌ | Packaging and Shipping of DS |
| ■ | Project Audit and Final Report |
Documentation:
We have a validated electronic Quality Management System, MasterControl, to automate and integrate processes for meeting quality standards and complying with regulatory requirements. Master Control manages documents, training and risk; processes and audits; and facility and equipment calibration and maintenance program. Documentation in MasterControl includes, but is not limited to, raw material specifications, product specifications, MPR, equipment operation SOPs, and analytical method SOPs and protocols.
CGMP manufacturing will be performed in compliance with CGMP regulations, including approved Master Batch Records, CGMP cell banks, active environmental monitoring and QA release of all raw materials and consumables. Raw materials will be purchased using QA-approved material specifications from QA-approved suppliers in accordance with our Supplier Selection, Assessment and Approval procedures (SOP-20-00018). CGMP runs will be performed in the manufacturing core using the same equipment, facilities and personnel as utilized for the Engineering Run.
Production:
Trained manufacturing personnel will execute one CGMP production lot in accordance with QA-approved production records and SOPs. The CGMP runs will be performed in the manufacturing core using the same equipment, facilities and personnel as utilized for the Engineering run. The manufacturing core features ISO 8 in-operation (Grade C) processing rooms for all closed system operations. All open manipulations will occur within the ISO 5 area (Grade A BSC) located within an ISO 7 in-operation (Grade B) suite adjacent to the main processing room. Samples will be taken throughout the manufacturing process according to the batch record and product specifications. The process will be executed aseptically from start to finish, as demonstrated in the process simulation as part of Task 7. Bulk DS will be stored at ≤ -70°C fill/finish at Ology Bio.
The CGMP material will be tested according to the DS specifications and tests previously defined. QC conducts in-process and lot release testing per SOPs and sampling plans. QA will provide the final review of batch records, environmental monitoring and analytical results. QA will also provide release of CGMP DS via a COA, ensuring that the DS product lot meets all technical specifications and is acceptable for use in GLP, nonclinical studies and Phase 1 clinical studies.
Ology Bio will perform limited stability testing as outlined in Task 5 and will provide samples as defined.
A list of deliverables for Task 6 is shown in Error! Reference source not found..
| Table 18. Task 6 Deliverables | |
| Task | Deliverable |
| Task 6: CGMP Run and Stability Testing | QA-Approved Batch Production Records for each of the two drug substance candidates |
| QA-Approved COA(s) (per lot, total of two) | |
| Stability testing samples, data and reports | |
| Campaign summary reports | |
| DS materials for DP formulation | |
Page 12 of 22
Task 7: Drug Product Engineering Run
Table 19. Task 7 Technical Assumptions
| Technical Assumption(s) |
| Two different drug products are required for this project |
| Maximum DP lot size is 2,000 single-dose vials per lot |
| Intended DP storage temperature is < -20°C |
| DP will include an adjuvant provided by Blue Water with the final formulations |
| DP process qualification runs will be required |
After completing the DS Engineering run, Ology Bio will execute an Engineering run of the DP filling process using the final container and closure method agreed upon with Blue Water (anticipated to be a 2 ▇▇ ▇▇▇▇) for each of the two Drug Products. The final formulation will include an adjuvant provided by Blue Water. DP vials will be tested according to developed release criteria (Table 20) and placed on limited stability studies (Table 21).
Table 20. Drug Product Release Assay
| Assay | Method | Location | Specification | |
| Physiochemical Properties | ||||
| Appearance | Visual Observation | Ology Bio | Clear, colorless liquid; no particles | |
| pH | USP<791> | Ology Bio | 7.4 ±0.3 | |
| Osmolality | USP<785> | Ology Bio | 250 – 350 mOsmol/kg | |
| Safety | ||||
| Endotoxin | USP<85> | Ology Bio | < 10 EU/dose; dose = 100 µg | |
| Sterility | 21CFT610.2 | Ology Bio | No growth ≥14 days | |
| Bioburden | Membrane Filtration | Ology Bio | < 10 CFU/mL | |
| General Safety | 21CFR610.11 | Ology Bio | Pass | |
| Content | ||||
| Protein Concentration | BCA | Ology Bio | Report | |
| Identity | ||||
| Western | Ology Bio | Identity Confirmed | ||
| Presence pf Blue Water Vaccine Candidate | ||||
| Purity | ||||
| Purity | SDS-PAGE | Ology Bio | >95% monomer | |
| Potency | ||||
| In vitro immunopotency | ELISA | Blue Water | TBD |
Page 13 of 22
Table 21. Drug Product Stability
| Test | Location | Acceptance Criteria | 0d | 1m | 3m | 6m | 9m | 12m | 18m | 24m | |
| Visual Inspection | Ology Bio | No alum – no particles observed | X | X | X | X | X | X | X | X | |
| Visual Inspection | Ology Bio | With alum – white substance with no colored particles | X | X | X | X | X | X | X | X | |
| pH | Ology Bio | 6.5 – 7.5 | X | X | X | X | X | X | X | X | |
| BCA | Ology Bio | TBD | X | X | X | X | X | X | X | X | |
| In vitro immunopotency | Blue Water | TBD | X | X | X | X | X | X | X | X | |
| SDS/PAGE/Western blot/densitometry to monitor protein degradation | Ology Bio | TBD | X | X | X | X | X | X | X | X | |
| Sterility | Ology Bio | No fungal or bacterial growth observed | X | X | X |
A list of deliverables for Task 7 is shown in Error! Reference source not found..
| Table 22. Task 7 Deliverables | ||
| Task | Deliverable | |
| DP Development Plan | ||
| DP Development Report | ||
| Non-CGMP Engineering Run DP vials suitable for use in preclinical studies | ||
| Task 7: DP Engineering Lots | DP Engineering Summary Report consisting of release testing results | |
| Media Fill Qualification Report | ||
| Up to 2,000 vials of Engineering DP per lot (two lots) | ||
| Stability study protocol | ||
| Stability study report | ||
Task 8: OPTIONAL – CGMP Drug Product
Table 23. Task 8 Technical Assumptions
| Technical Assumption(s) |
| Two different drug products are required for this project |
| Maximum DP lot size is 2,000 single-dose vials per lot |
| Intended DP storage temperature is < -20°C |
| DP will include an adjuvant provided by Blue Water with the final formulations |
| DP process qualification runs will be required |
In accordance with FDA Guidance for Industry, “Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice,” Sept 2004, aseptic formulation and fill validation (media fill validation) will be conducted using a maximum of 2,000 vials per lot in a mutually approved container/closure system (vial, stopper, seal). Three consecutive successful media fill Validation Runs will be performed using TBS to simulate the formulated DP according to an approved media fill validation batch record. In addition, appropriate interventions (extended processing times, simulation of spillage and clean-up of spillage, changing out of the fill needle) will be incorporated into the validation activities.
Page 14 of 22
Phase 1 DP formulation and liquid product fill operations will be conducted at Ology Bio. Using QA-approved production documentation, the DS lot will be formulated to achieve the final concentration of the to-be-determined titer in the selected final formulation (determined by Blue Water). Formulated product will be filled into the mutually approved container/closures within an aseptic area. A maximum of 2,000 vials per DP lot will be targeted for filling. QC will conduct lot release testing as summarized in Error! Reference source not found. per batch records and sampling plans. QA will provide the final review and release, ensuring that the DP lots meet all technical specifications and are acceptable for use in GLP nonclinical studies and clinical studies. Stability testing of CGMP DP lots is described in Error! Reference source not found..
A list of deliverables for Task 7 is shown in Table 24.
| Table 24. Task 8 Deliverables | ||
| Task | Deliverable | |
| Up to 2,000 vials of CGMP DP per lot (two lots) | ||
| COAs for released CGMP DP | ||
| Task 7: DP CGMP Lots | QA-approved batch production records (per lot, two lots) | |
| QA-approved Campaign Summary Report | ||
| Stability study protocol | ||
| Stability study report | ||
Task 9: Regulatory Support for Preclinical IND-Enabling Studies and IND Preparation
Table 25. Task 9 Technical Assumptions
| Technical Assumption(s) |
| Regulatory support for two different drug substance candidates and two drug product candidates is required |
| Non-CGMP Engineering lot will be used for the toxicity study |
| Ology Bio assumes that clinical SMEs will be provided by the Sponsor to support protocol development and review |
| Electronic Publishing costs are not included in this proposal |
Subtask 9.1: Pre-Clinical Tox Study
Ology Bio will support Blue Water in the development of a nonclinical safety plan to support IND filing. Based on Ology Bio Regulatory experience, CBER/FDA expects high-quality of material for the IND-enabling toxicity studies that is either CGMP material or comparable to CGMP material. To reduce risk, the schedule linked this study to the CGMP lot. Blue Water can consider risks as it reviews the nonclinical safety plan.
Ology Bio’s Nonclinical SME will work with the subcontractor, IITRI to develop a protocol for an IND-enabling study based on feedback received from the FDA during the Pre-IND meeting (Subtask 8.2). Ology Bio will oversee the performance and of a GLP-Compliant Repeat Dose Toxicity Study of the Influenza Vaccine Candidate in Rabbits. The objective of the study will be to determine the immunogenicity, target organ toxicity, and reversibility of the influenza vaccine in rabbits following a repeat dosing regimen to support a Phase 1 clinical study.
Subtask 9.2: Pre-IND Meeting Support
Ology Bio will support a Type B Pre-IND Meeting to ensure successful entry into first-in-man studies. Effective communication with the FDA during the pre-IND stage of product development fosters a strong working relationship and is important for clearance of the IND. Ology Bio Regulatory Affairs (RA) team will respond to information requests received prior to the meeting, support meeting participation and prepare meeting minutes.
Page 15 of 22
Our RA team will be engaged in practice sessions to develop responses to potential FDA questions and address concerns to avoid delays in product development. Our RA team tracks risks associated with entry into clinical development and ensures that the meeting reduces risk by proper preparation.
The Pre-IND meeting will include briefing materials describing the Phase 1 Protocol Synopsis; nonclinical toxicity plan; CMC technical information including cell banking and detailed manufacturing process descriptions; release; and stability information. Specific questions will focus on acceptability of the information to be provided in the IND.
Subtask 9.3: Regulatory Technical Writing
To support Blue Water in developing their IND application, our RA experts will prepare CMC sections for Blue Water’s vaccine. Ology Bio will provide Tier 1, Tier 2 and Tier 3 regulatory support, which includes technical review of strategic documents (i.e., specifications, change controls, protocols, risk assessments and technical reports) and CMC technical writing. Ology Bio will author the DS CMC information (Quality Modules 3.2.S, 3.2.P and 3.2A) in ICH Common Technical Document (CTD) format, delivered as Microsoft Word documents. CMC technical writing will be limited to manufacturing and testing activities managed by Ology Bio, and placeholders will be included for Blue Water-managed activities. In addition, Ology Bio will support development of Modules 4 and 5 with deliverables provided in Word. Electronic Publishing costs are not included in this proposal.
The Ology Bio RA team is responsible for managing, writing, completing or editing all technical writing assignments; obtaining drop-in documents from the SMEs; assembling all documents and forms into a submission package; and uploading the submission for review and approval. Our RA team works with SMEs as needed to complete editing and addressing reviewers’ comments. RA is responsible for working with QA staff to ensure that all information/data has been reviewed for accuracy prior to Client review of documents. For this effort, Ology Bio assumes that clinical SMEs will be provided by the Sponsor to support protocol development and review.
Our RA team uses eCTD Word templates that provide authors with the ability to create documents that adhere to a single standard for consistency to the FDA and to our clients. Scientifically sound and accurate CMC writing to support CTD Module 3 development is critical to the success of the CMC communications with Regulatory authorities. Cost and regulatory operations support for electronic publishing and filing of the IND is not included.
Table 26. Task 9 Deliverables
| Task | Deliverables |
| Preclinical study plan development (PK and toxicity studies) | |
| Preclinical Protocol Drafts | |
| Task 8: Regulatory Support for Preclinical IND- | Module 3 for CGMP DS and DP |
| Enabling Studies and IND Preparation | IND support documentation (MS Word Deliverables) |
| Clinical trial documentation to support Phase 1 clinical study | |
| (Investigator Brochure and Phase 1 Protocol) |
Page 16 of 22
3. PROJECT SCHEDULE
The project schedule is presented in Table 27. The proposed start date for this project is September __, 2019; this start date is subject to change based on the date this proposal is accepted and signed and availability of the facility.
Table 27. Project Schedule
| Task | Description | Start | End |
| 1 | Technology Transfer and Process Establishment | Oct 2019 | Jan 2020 |
| 2 | Analytical Assay Development | Oct 2019 | Feb 2020 |
| 3 | CGMP Master and Working Cell Banking, Stability | Nov 2019 | Feb 2021 |
| 4 | Process Development and Scale-up | Jan 2020 | Jun 2020 |
| 5 | Engineering Run and Stability Testing | Jun 2020 | Aug 2021 |
| 6 | OPTIONAL: CGMP Run and Stability Testing | Aug 2020 | Oct 2021 |
| 7 | DP Engineering Lot | Jun 2020 | Sep 2021 |
| 8 | OPTIONAL: CGMP DP Lot | Sep 2020 | Oct 2021 |
| 9 | Regulatory Support for Preclinical IND-Enabling Studies and IND Preparation | Jul 2020 | Jul 2021 |
4. PROJECT BUDGET
Table 28. Project Budget
| Task | Description | Task Price1 | Pass-Through Costs2 | Total with Estimated Pass-Through |
| 1 | Technology Transfer and Process Establishment | $171,000 | $6,000 | $177,000 |
| 2 | Analytical Assay Development | $225,000 | $27,000 | $252,000 |
| 3 | CGMP Master & Working Cell Banks | $306,000 | $230,000 | $536,000 |
| 4 | Process Development and Scale-Up | $324,000 | $413,000 | $737,000 |
| 5 | Engineering Runs and Stability Testing | $444,000 | $347,000 | $791,000 |
| 6 | OPTIONAL: CGMP Runs and Stability Testing | $594,000 | $489,000 | $1,083,000 |
| 7 | DP Engineering Lot and Stability Testing | $357,000 | $79,000 | $436,000 |
| 8 | OPTIONAL: CGMP DP Lot and Stability Testing | $393,000 | $84,000 | $477,000 |
| 9.1 | Pre-Clinical Tox Study3 and Protocols | $166,000 | $325,000 | $491,000 |
| 9.2 | Pre-IND Meeting Support | $253,000 | $0 | $253,000 |
| 9.3 | Regulatory Technical Writing | $333,000 | $0 | $333,000 |
| TOTAL – with optional tasks | $3,566,000 | $2,000,000 | $5,566,000 | |
| TOTAL – optional tasks (6 & 8) | $987,000 | $573,000 | $1,560,000 | |
| TOTAL – without optional tasks | $2,579,000 | $1,427,000 | $4,006,000 | |
| 1 | Task prices are based on estimated time. |
| 2 | Material costs are estimated for budgeting purposes and include a 15% material handling fee. |
| 3 | GLP compliant IND-enabling tox study performed by Ology Bio subcontractor |
Page 17 of 22
5. PAYMENT SCHEDULE
Table 29. Payment Schedule
| Payment Table | ||||
| Milestone | Task | Description | Deliverables | Payment |
| MS1 | 1 – 4 | Initiation of Tasks 1 – 4 (50%) | $513,000 | |
| MS2 | 1 | Technology Transfer and Process Establishment |
● Final Schedule Agreement ● Project Management Plan ● Project Charter Process Development Plan ● Process Establishment Plan and Report ● Preliminary ▇▇▇▇ of Materials |
$85,500 |
| MS3 | 2 | Analytical Assay Development |
● Analytical Assay Qualification Plan ● Approved Qualification Reports |
$112,500 |
| ● In-Process and Release Assay Specifications | ||||
| MS4 | 3 | CGMP Master & Working Cell Banks | ● Draft MCB Master Batch Record | $153,000 |
|
● Executed MCB Master Batch Record ● Draft WCB Master Batch Record ● Executed WCB Master Batch Record ● Approved Stability Study Plan for MCB ● Approved Stability Study Plan for WCB ● Minimum of 300 cGMP MCB Vials ● Minimum of 300 cGMP WCB Vials | ||||
| MS5 | 4 | Process Development and Scale-Up |
● Approved Process Development Report ● Process Development Run Materials ● Approved Process Scale-Up Final Report ● Scale-Up Run Materials ● Engineering Run Process Parameters ● Technology Transfer Protocol ● Draft Batch Records ● Sampling Plans ● Confirmation Run Report ● Confirmation Run Materials |
$162,000 |
| MS6 | 5 | Initiation of Task 5 (50%) | $222,000 | |
| MS7 | 5a Engineering Run |
● DS Engineering Report ● DS Engineering Stability Protocol ● Finalized BOM ● Finalized CGMP Batch Record Templates ● Finalized CGMP DS Specifications ● Engineering Non-CGMP DS COA MSDS ● Updated Tech Transfer Protocol (if needed) ● Engineering Non-CGMP DS Product |
$205,350 | |
| MS8 | 5b Engineering Run Stability Testing | ● DS Engineering Stability Report | $16,650 | |
Page 18 of 22
| MS9 | 7 | Initiation of Task 7 (50%) | $178,500 | |
| MS10 | 7a Engineering Drug Product |
● DP Development Plan ● DP Development Report ● Non-CGMP Engineering Run DP Vials Suitable Preclinical Studies ● DP Engineering Summary Report |
$89,250 | |
| MS11 | 7b Engineering Drug Product Stability Testing (12 months) |
● Stability Study Protocol ● Interim Stability Study Report |
$44,625 | |
| MS12 | 7c Engineering Drug Product Stability Testing (24 months) | ● Final Stability Study Report | $44,625 | |
| MS13 | 9.1 | Initiation of Pre-Clinical Tox Study and Protocols | $83,000 | |
| MS14 | Completion of Pre-Clinical Tox Study and Protocols | ● Preclinical Study Plan Development | $83,000 | |
|
(PK and Toxicity Studies) ● Preclinical Protocol Drafts | ||||
| MS15 |
9.2
9.3 |
Initiation of Pre-IND Meeting Support | $126,500 | |
| MS16 | Completion of Pre-IND Meeting Support |
● Module 3 for CGMP DS and DP ● IND Support Documentation (MS Word Deliverables) |
$126,500 | |
| MS17 | Initiation of Regulatory Technical Writing | $166,500 | ||
| MS18 | Completion of Regulatory Technical Writing | ● Clinical Trial Documentation to Support Phase 1 Clinical Study | $166,500 | |
| Labor Total | $2,579,000 | |||
| Estimated Consumable Pass-Through Costs | $1,427,000 | |||
| Estimated Overall Total | $4,006,000 | |||
Page 19 of 22
Table 30. Optional Tasks Payment Schedule
| Payment Table (Optional Tasks) | ||||
| Milestone | Task | Description | Deliverables | Payment |
| MS19 | 6 | Initiation of Task 6 (50%) | $297,000 | |
| MS20 | 6a CGMP Run |
● QA-Approved Batch Production Records ● QA-Approved COA(s) (per lot) ● Stability Testing Samples ● Campaign Summary Report ● DS Materials for DP Formulation |
$282,150 | |
| MS21 | 6b CGMP Run Stability Testing |
● Stability Testing Data ● Stability Testing Reports |
$14,850 | |
| MS22 | 8 | Initiation of Task 8 (50%) | $196,500 | |
| MS23 | 8a CGMP Drug Product |
● Media Fill Qualification Report ● Up to 2,000 Vials of CGMP DP |
$98,250 | |
|
● COAs for Released CGMP DP ● QA-Approved Batch Production Records ● QA-Approved Campaign Summary Report | ||||
| MS24 | 8b CGMP Drug Product Stability Testing (12 months) |
● Stability Study Protocol ● Interim Stability Study Report |
$49,125 | |
| MS25 | 8c CGMP Drug Product Stability Testing (24 months) | ● Final Stability Study Report | $49,125 | |
| Labor Total | $987,000 | |||
| Estimated Consumable Pass-Through Costs | $573,000 | |||
| Estimated Overall Total | $1,566,000 | |||
6. MATERIALS
Materials will be invoiced to Blue Water as per Section 3.3 of the MSA.
7. PAYMENT TERMS
Payment terms are defined in Section 6.4 of the MSA.
8. STORAGE FEES
$350 / month / freezer or refrigerator shelf
$1,200 / month / entire freezer or refrigerator
Page 20 of 22
9. GENERAL ASSUMPTIONS:
The schedules, estimates and costs contained within this Project Addendum are based on the Listing of Technical Assumptions and the following general assumptions.
1. Ology Bio Technical Approach is a suggested pathway based on the information provided in the Blue Water Request for Proposal (RFP). Additional and/or replacement of the techniques as a result of new and/or more (or less) detailed information from Blue Water may affect the content and pricing.
2. Information or issues discovered after contract award through the gap analysis performed on the technical transfer package provided to Ology Bio, or through analytical and/or process development activities, may require changes to proposed scope and costs. If such situations arise, Ology Bio will propose these changes to Blue Water through a formal Change Request which will require Blue Water approval before the changes can proceed.
3. Blue Water will make available the appropriate subject matter experts and stakeholders as needed.
4. Blue Water will provide sufficient materials required to begin assay development.
5. Blue Water assumes that active and responsive participation and availability by Blue Water SMEs, stakeholders, etc., will exist throughout the length of this project in support of project scope, schedule, and team.
6. Access to development/practice/test documents will be available at contract start.
7. Full cooperation and conditions obtained from any/all applicable external third parties (manufacturers, service providers, leasers, etc.) required by the scope of this project will be acceptable to Ology Bio. Unfavorable conditions (terms, costs, etc.) will require that alternative solutions be found.
8. Timelines are bound to the specific period outlined in this Project Addendum. As such, the appropriate space and resources will be allocated to this project during that timeframe. In the event of delays resulting in activity or inactivity of Blue Water, additional charges and an extension of the timeline may become necessary.
9. Ology Bio will work in good faith based on agreed upon terms and in cooperation of the needs of Blue Water. All activities associated with this project remains at the discretion of Ology Bio.
10. A mutually agreed-upon Decision Log will be used to make and record non-substantial changes/modifications to the contract without the need for a complete formal amendment. The Decision Log will be referenced as incorporated into the contract.
10. MSA TERMS:
All of the terms and conditions set forth in the MSA, to the extent not expressly modified herein, are hereby incorporated into this Project Addendum as if set out in full herein. If any terms in this Project Addendum conflict with the terms of the MSA, the terms in the MSA will govern, except as specifically modified herein in accordance with the MSA. All capitalized terms not otherwise defined herein shall have the meanings ascribed to such terms in the MSA.
Page 21 of 22
IN WITNESS WHEREOF, the parties have caused this Project Addendum to be executed by their duly authorized representatives.
| Ology Bioservices, Inc. | Blue Water Therapeutics, Inc. | |||
| By: | /s/ ▇▇▇▇▇▇▇ ▇▇▇▇▇ | By: | /s/ ▇▇▇ ▇▇▇▇▇▇▇▇▇ | |
| Name: | ▇▇▇▇▇▇▇ ▇▇▇▇▇ | Name: | ▇▇▇ ▇▇▇▇▇▇▇▇▇ | |
| Title: | Title: | |||
Page 22 of 22