AGREEMENT
Exhibit 10.16
AGREEMENT
THIS AGREEMENT is made and entered into as of this 24 day of January, 2010 by and between Vascular Biogenics Ltd., a company registered under the laws of the State of Israel (“VBL”) and Xxxx. Xxxxx Xxxxxx (“Xxxxxx”);
| WHEREAS: | Xxxxxx is a co-founder and employee of VBL, currently serving as VBL’s Chief Scientific Officer pursuant to that certain employment agreement between the parties dated March 2001 and as amended (the “Employment Agreement”). In addition, Xxxxxx serves as a senior cardiac researcher at the Tel Aviv Sourasky Medical Center (“Sourasky”); |
| WHEREAS: | The parties acknowledge that the engagement by VBL and Sourasky in parallel may cause uncertainty in connection with the ownership of certain inventions developed by Xxxxxx during the course of his engagement and related intellectual property; and |
| WHEREAS: | The parties wish to clarify the matter and set the understandings as between the parties according to the terms hereof; |
NOW, THEREFORE, THE PARTIES AGREE AS FOLLOWS:
| 1. | Intellectual Property Rights |
| 1.1 | Other than as specifically indicated in Appendix A, all right, title and interest in and to the intellectual property developed, conceived or reduced into practice by Xxxxxx since the commencement of Xxxxxx engagement with VBL belong exclusively to VBL in accordance with the terms of Section 6 of the Employment Agreement (“VBL IP”). The parties further believe that Sourasky has no or will not have any claim, basis for a claim, right or interest in and to the VBL IP. |
| 1.2 | VBL hereby represents and warrants that it does not have any claim or right in and to the intellectual property specified in Appendix A or otherwise derived or resulting therefrom (“Other IP”) and hereby irrevocably waives any right in connection therewith. In the event that VBL is found to hold any rights in the Other IP, it hereby irrevocably sells, assigns, conveys and transfers to Xxxxxx, his assigns and successors all such right, title and interest anywhere in the world in and to such Other IP. |
| 1.3 | VBL agrees to execute upon the request of such additional instruments, applications, declarations and forms, as may be necessary under any relevant law or as may be required by any official or authority, to continue, secure, defend, register and otherwise give full effect to, and perfect the foregoing assignment. |
| 1 | 16-01\01840495 |
| 1.4 | Xxxxxx agrees to support the position and VBL rights as set froth in Section 1.1 above in his correspondences with Sourasky and to assist VBL in any manner in connection with protecting VBL rights in the VBL IP against any claim made by Sourasky, if any. |
| 2. | Revenue Sharing |
In the event that either Sourasky or Xxxxxx commercializes or provides any third party with the right to commercialize or otherwise exploit the Other IP or any other intellectual property if it is later found to be owned by Sourasky due to the engagement of Xxxxxx therewith, regardless of the parties agreement in Section 1 above, and as a result of such commercialization Xxxxxx receives any benefit, right or distribution (whether in cash or in kind), then Xxxxxx undertakes to transfer or ensure the receipt of 50% of such benefit, right or distribution by VBL within 30 days following the receipt thereof. In the event that the transfer of such portion of the benefit, right or distribution is not feasible, the parties shall agree on an appropriate mechanism to ensure VBL rights therein.
| 3. | General Provisions. |
| 3.1 | Governing Law; Arbitration. This Agreement shall be governed by and construed in accordance with the laws of the State of Israel. In the event that a settlement to a dispute hereunder cannot be reached within thirty (30) days, either party may request that such controversy be finally settled by arbitration pursuant to the Arbitration Law, 1968, by one arbitrator appointed by the Chairman of the Israeli Bar Association and acting in accordance with such law. The arbitrator will not be bound by rules of evidence or procedure and will give the reasons for his judgment. The arbitrator’s decision shall be final and enforceable in any court. This paragraph constitutes an agreement to arbitrate for purposes of the Arbitration Law, 1968. |
| 3.2 | Entire Agreement. This Agreement, and all schedules attached hereto, constitutes the entire agreement of the parties with respect to the subject matter hereof. This Agreement may be amended only by a document executed by all parties purporting to effect such an amendment. |
| 3.3 | Assignment. Neither party may assign or transfer its rights and obligations under this Agreement without the prior written consent of the other party, provided, however, that VBL may, without such consent, assign this Agreement and the rights, obligations and interests hereunder, in whole or in part, to any purchaser of all or substantially all of its assets or to any successor corporation resulting from any merger or consolidation. |
| 3.4 | Non-Waiver. A failure by a party hereto to exercise or enforce any rights conferred upon it by this Agreement shall not be deemed to be a waiver of any such rights or operate so as to bar the exercise or enforcement thereof at any subsequent time or times. |
| 2 | 16-01\01840495 |
| 3.5 | Notice. For the purpose of this Agreement, notices and all other communications provided for in the Agreement shall be in writing and shall be deemed to have been duly given when personally delivered or sent by registered mail, postage prepaid, addressed to the respective addresses last given by each party to the other. All notices and communications shall be deemed to have been received on the date of delivery thereof, except that notice of change of address shall be effective only upon receipt. |
{SIGNATURE PAGE FOLLOWS}
IN WITNESS WHEREOF the parties have caused this Agreement to be executed as of the day first written above.
| VASCULAR BIOGENICS LTD. | ||||||||
| by: | /s/ Xxxx Xxxxxx | /s/ Xxxxx Xxxxxx | ||||||
| name: | Xxxx Xxxxxx | XXXX. XXXXX XXXXXX | ||||||
| title: | CEO | |||||||
| 3 | 16-01\01840495 |
Appendix A
Monoclonal antibodies targeting Eotaxin and Eotaxin receptors as therapeutic agents.
| 4 | 16-01\01840495 |
Eotaxin-2 (CCL24) inhibitors in inflammatory, autoimmune, and cardiovascular disorders
FIELD OF INVENTION
The present invention concerns the use of inhibitors of eotaxin-2 (CCL11) in the treatment of inflammatory, autoimmune, and cardiovascular disorders, in particular anti eotaxin-2 polyclonal or monoclonal antibodies.
BACKGROUND OF INVENTION
Chemokines are small cytokines which act as chemoattractants for leukocytes, coordinating both homeostatic trafficking of these cells as well as recruiting specific cell populations to sites of inflammation. Chemokine dysregulation is considered to play a part in a wide spectrum of human disease involving the immune system including inflammation and and autoimmunity (1).
The chemokine eotaxin-2 (also termed CCL11) is a potent chemo attractant for inflammatory cells. Eotaxin-2 binds the eosinophil receptor CCR3 and possesses a potent chemotactic activity for eosinophils (2-4), basophils (4), and Th2-type lymphocytes (5). There is abundant data on the pleiotropic effects of this chemokine. Eotaxin-2 is expressed in various types of endothelial cells (5-9), and induces angiogenic and migratory responses in endothelial (10) and smooth muscle cells (11).
Two additional cytokines having properties similar to eotaxin-2 (termed “eotaxin” and “eotaxin-3”) have been identified in humans. Eotaxin-2 is only 39% homologous to eotaxin, and the two polypeptides differ almost completely in the
| 5 | 16-01\01840495 |
NH2-terminal region (12). Eotaxin-2 is located on chromosome 7q11.23 and eotaxin is located on chromosome 17q21.1. The eotaxin-3 gene lies close to the eotaxin-2 gene on chromosome 7 but shares only 33% homology with it. All these chemokines bind specifically to the CCR-3 receptor. CCR3, the eotaxin receptor, is a 7-transmembrane G protein-coupled receptor which, beside being expressed by eosinophils, is expressed by a wide array of cell types including macrophages and endothelial cells (13).
WO 97/00960 discloses nucleic acids which encode human eotaxin, as well as isolated or recombinant human eotaxin proteins. WO 97/00960 also discloses methods of use of the eotaxin proteins in the recruitment of eosinophils to a particular site or in the treatment of allergic conditions.
CCR3 expression was originally extensively studied in the pathogenesis of asthma and allergy, where it continues to serve as a therapeutic target (14). More recently however, a role for this pathway has emerged in the study of additional inflammatory and autoimmune disorders including inflammatory bowel disease (15), multiple sclerosis (16) and rheumatoid arthritis (RA).
Rheumatoid arthritis (RA) is a common, chronic inflammatory disease, characterized by intense, destructive infiltration of synovial tissue by a broad spectrum of inflammatory cells (17). Multiple cytokines, derived from macrophages and fibroblasts are responsible for the secretion of both cytokines and chemokines in (RA)(18). The accumulation of leukocytes in the joint space leads to secretion of tissue degrading factors, including cytokines and matrix degrading enzymes.
| 6 | 16-01\01840495 |
Chemokine inhibition has previously been tested as a therapeutic option in adjuvant induced arthritis, a commonly used animal model of RA (19). Using the same model, CCR3 has been shown to play a role in recruitment of leukocytes to synovial tissue (20). Differential expression of many chemokines and chemokine receptors has also been demonstrated in serum and synovial tissue of RA patients (21).
Inflammation with involvement of cytokines and chemokines is thought to play a pivotal role also in promoting atherosclerotic plaque growth and propensity to destabilize and subsequently rupture (22, 23). Eotaxin/CCL24 receptor (CCR3) is expressed in plaque macrophages (24). A clinical study demonstrated that in a cohort of healthy men, a non-conservative polymorphism in the eotaxin gene has been associated with increased risk for myocardial infarction (25). In a subsequent study, it has been found that increased circulating eotaxin level is associated with the presence of coronary atherosclerosis and ischemia (26, 27).
Atherosclerosis is a process in which fat deposition progresses in the arterial wall leading to progressive narrowing of the lumen. The mature plaque is composed of two basic structures: the lipid core and the fibrous cap. The smaller the lipid core and the thicker the fibrous cap, the more stable the plaque is, meaning that its propensity to rupture and cause myocardial infarction or unstable angina are increased. It is now clear that most plaques that cause acute coronary syndromes (e.g., myocardial infarction and unstable angina) are angiographically shown to have <70% stenosis (reviewed in 28, 29). Approximately 60% of these lesions are caused by rupture of plaques with a large thrombogenic core of lipid and necrotic debris (including foci of macrophages, T cells, old hemorrhage, angiogenesis, and calcium). The ruptured cap is thin, presumably because macrophages secrete matrix metalloproteinases that digest it as they move across plaque, and because smooth muscle cells (the supporting element of the plaque) are depleted due to senescence or apoptosis caused by several factors, such as inflammatory cytokines.
| 7 | 16-01\01840495 |
WO 06/93932 discloses methods for the detection or diagnosis of atherosclerosis by measuring the level of eotaxin in an individual’s serum. The application further suggests that detection of elevated eotaxin levels in serum may provide a means to diagnose atherosclerosis prior to the onset of symptoms.
None of the above publications teach or suggest eotaxin-2 as a target for therapeutic intervention for the treatment of autoimmune or cardiovascular diseases.
SUMMARY OF THE INVENTION
The present invention is based on the finding that inhibition of eotaxin-2 by polyclonal or monoclonal antibodies, has a significant protective effect in animal models of inflammatory diseases such as rheumatoid arthritis, experimental autoimmune encephalomyelitis (EAE), colitis, diabetes, and atherosclerosis. Without wishing to be bound by theory, the protective effects could be mediated, at least in part, by attenuation of the adhesive and migratory properties of the active inflamatory cells (lymphocytes and mononuclear cells). The present invention thus introduces eotaxin-2 as a novel target for developing therapeutics to treat inflammatory and/or autoimmune disorders. The invention also provides specific anti-eotaxin 2 antibodies for use alone or in combination with other therapeutic agents in the treatment of such disorders.
Inflammatory and/or autoimmune diseases include, for example, psoriasis, inflammatory bowel disease (ulcerative colitis and Crohn’s disease), rheumatoid arthritis, diabetes, multiple sclerosis, systemic lupus erythematosus (SLE), scleroderma and pemphigus.
| 8 | 16-01\01840495 |
Accordingly, by a first of its aspects, the present invention provides a pharmaceutical composition for treating inflammatory or autoimmune diseases comprising an Eotaxin-2 antagonist and a pharmaceutically acceptable carrier or excipient.
In one embodiment said Eotaxin-2 antagonist is an anti Eotaxin-2 antibody.
The anti-eotaxin-2 antibody may be a monoclonal antibody or a polyclonal antibody. The anti-eotaxin-2 antibodies may be human antibodies, humanized antibodies or chimeric antibodies.
In certain specific embodiments, said anti-eotaxin-2 antibody is a monoclonal antibody secreted by hybridoma G7, or hybridoma G8. In another embodiment, the Eotaxin-2 antagonist is an antisense or a siRNA molecule directed against Eotaxin-2 mRNA.
In another embodiment, the Eotaxin-2 antagonist is a small molecule.
By another aspect, the present invention provides a method for treating inflammatory or autoimmune diseases comprising administering to a patient in need thereof an eotaxin-2 antagonist or the pharmaceutical composition of the invention.
In accordance with certain embodiments of the invention, said autoimmune disease is selected from the group consisting of rheumatoid arthritis, inflammatory bowel disease, colitis, and diabetes.
By yet another aspect, the present invention provides a method for inhibiting atherosclerotic plaque formation comprising administering to a patient in need thereof an Eotaxin-2 antagonist or the pharmaceutical composition of the invention.
| 9 | 16-01\01840495 |
The invention also provides a method for stabilizing an atherosclerotic plaque comprising administering to a patient in need thereof an eotaxin-2 antagonist or the pharmaceutical composition of the invention.
The invention also provides a method for preventing major cardiovascular events in a patient with acute coronary syndrome comprising administering to said patient an eotaxin-2 antagonist or the pharmaceutical composition of the invention.
The term major cardiovascular events encompasses but is not limited to, ST- or NON-ST elevation, myocardial infarction, unstable angina and new onset angina.
The methods of the invention also encompass administration of said eotaxin-2 antagonist or said pharmaceutical composition in combination with at least one additional therapeutic agent.
In accordance with certain embodiments said at least one additional therapeutic agent is selected from a group consisting of chemotherapeutics, cytokines, peptides, antibodies and antibiotics.
For the treatment of Arthritis, the at least one additional therapeutic agent for administration in combination with said eotaxin-2 antagonist or said pharmaceutical composition includes, methotrexate, a steroid, anti-TNFa antibodies, anti-IL6 R antibodies, or anti-CD20 antibodies.
For the treatment of colitis, the at least one additional therapeutic agent for administration in combination with said eotaxin-2 antagonist or said pharmaceutical composition includes, but is not limited to, cyclosporine, NSAIDS (non-steroidal anti inflammatory drugs), or steroids.
| 10 | 16-01\01840495 |
For the treatment of multiple sclerosis, the at least one additional therapeutic agent for administration in combination with said eotaxin-2 antagonist or said pharmaceutical composition includes, but is not limited to, copaxone, interferon-beta, IVIG, or a monoclonal antibody to VLA-4 (e.g. Tysabri).
In accordance with one embodiment of the invention said at least one additional therapeutic agent is administered simultaneously with the eotaxin-2 antagonist or said pharmaceutical composition.
In accordance with another embodiment of the invention said at least one additional therapeutic agent and said eotaxin-2 antagonist or said pharmaceutical composition are administered sequentially.
In another aspect, the present invention provides a hybridoma cell line secreting an anti-eotaxin 2 monoclonal antibody, wherein said hybridoma is selected from the group consisting of D8, G7 and G8.
The present invention also provides a monoclonal antibody directed against eotaxin-2, or any fragment thereof which retains the binding ability to eotaxin 2, wherein said monoclonal antibody is secreted from hybridoma D8, G7 or G8.
In another aspect, the present invention also provides use of eotaxin-2 antagonists in the treatment of inflammatory and autoimmune diseases. In one embodiment said eotaxin-2 antagonists are antibodies.
The invention also encompasses use of eotaxin-2 antagonists in the preparation of pharmaceutical compositions for treatment of inflammatory and autoimmune diseases. In one embodiment said eotaxin-2 antagonists are antibodies.
| 11 | 16-01\01840495 |
BRIEF DESCRIPTION OF THE DRAWINGS
In order to understand the invention and to see how it may be carried out in practice, embodiments will now be described, by way of non-limiting example only, with reference to the accompanying drawings, in which:
Detailed description of the embodiments
MATERIALS AND METHODS
Production of monoclonal antibodies
Several clones of mAbs were produced according to standard protocols. In short, Balb/C mice were immunized with 20µg of eotaxin-2 (Peprotec, USA) followed by four additional boosts. After confirming the presence of polyclonal anti-eotaxin-2 Abs in the sera, mice were sacrificed, cells were isolated from their spleens and hybridized with an NS/0 myeloma line, followed by clonal screening for binding to eotaxin-2. The hybridomas were then grown in serum-free media for 2-3 weeks, media collected and concentrated by 100 kDa centricons (Biological Industries, Israel). Cross-reactivity of one of the mAbs (D8) with murine eotaxin-2 was confirmed by ELISA.
Binding assays
Plates were coated with 1µ/ml of either eotaxin or eotaxin-2 (in buffer Carbonate), overnight at 4°C. The plates were washed with PBS-t 3 times and blocked with 2%BSA for 45 minutes at 37°C. Anti eotaxin-2 antibodies (the D8 clone) was put in serial dilutions in PBS for 1.5 hour at 37°C. Washing was repeated as indicated above and the plates were incubated for 1 hour at 37°C with a goat anti-mouse peroxidase conjugated antibody. Washing was repeated as above and binding was detected using a colorimetric substrate.
Splenocyte adhesion assays
In adhesion assays, C57Bl mice and Xxxxx rat splenocytes were separated on ficoll gradient and plated in 10cm dishes for an overnight incubation. On the next day cells were harvested and pretreated with increasing concentrations of D8 or total mouse IgG (5-50ug/ml) for two hours with rotation. Cells were then centrifuged and plated
| 12 | 16-01\01840495 |
on 96-well plates pre-coated with fibronectin. After one hour-incubation, non-adherent cells were washed away and the amount of adherent cells was analyzed using XTT kit (Biological Industries, Israel). Similar adhesion assays were performed using peripheral blood mononuclear cells (PBMCs) collected from healthy donors.
Migration assays
C57BL/6J-derived splenocytes, as well as rat splenocytes and human PBMCs pretreated with D8 (30ug/ml) were plated onto the upper chamber of a Trans-Well system. The lower chamber contained serum-free media supplemented with VEGF (vascular endothelial growth factor) (20ng/ml). Four hours later the media in the lower chamber was collected and cells counted using flow cytometry (number of cells collected per minute).
Plaques and cardiovascular in vitro studies
Human carotid plaque preparation and protein array
Human atherosclerotic plaques were recovered from two groups of patients. Stable plaques (n-4) were obtained from endarterectomy specimens of patients with asymptomatic severe carotid atherosclerosis. Representative of unstable plaques were specimens obtained upon percutaneous coronary angioplasty of culprit vessels from patients with acute myocardial infarctions (n=4). Thrombectomies were performed by dedicated suction devices. The obtained material consists of red thrombi, white thrombi and fragments of vulnerable plaques from the culprit artery. After washing and lysis, the remaining tissue comprises predominantly fragments of atherosclerotic ruptured plaques.
The RayBio™ Human Inflammation Antibody Array 3.1 (Ray Biotech, USA) was used for detection of 40 cytokines, chemokines and growth factors in stable and vulnerable human plaques. Briefly, plaques were homogenized in lysis buffer provided within the kit using pellet pestle. Arrays were incubated with 500 mg of protein of each sample and developed following the manufacturer’s instructions. The results were analyzed using XXXX 2.0 program.
| 13 | 16-01\01840495 |
Capillary cells
Murine capillary cells (H5V) were cultured in DMEM F-12 (Biological Industries, Israel) supplemented with 10% fetal calf serum (FCS) (Invitrogen) and 1% penicillin/streptomycin sulfate (Biological Industries, Israel). The cells were maintained at 37°C in a humid incubator with 8% CO2. Monocytoid U937 cells were cultured in complete medium RPMI 1640, containing 10% FCS and 1% penicillin/streptomycin sulfate at a concentration of 106cells/ml. The cells were maintained at 37°C in 5% CO2 humid incubator.
Adhesion assay with endothelial cells
In adhesion assays, H5V mouse endothelial cells were incubated for 72h in presence of oxLDL, 1ug/ml (prepared as previously described, 33). Then the cells were plated overnight in 96 well plates at concentration 4X103 cells/well in presence of oxLDL, 1µg/ml. On the next day, the cells were pretreated with the neutralizing goat anti mouse Eotaxin-2 antibody (Cytolab, USA) or goat IgG for one hour before U937 cells or spleen-derived lymphocytes from ApoE mice were added at a concentration of 8 X104 /well. After one hour-incubation, non-adherent cells were washed away and the amount of adherent cells was analyzed using XTT kit (Biological Industries, Israel).
PCR analysis of aortas and H5V endothelial cells
RNA from tissue samples and H5V endothelial cells was isolated by the guanidinium thiocyanate and phenol chloroform method using EZ-RNA kit (Biological Industries, Israel) following the manufacturer’s protocol. RT-PCR was carried out using AMV reverse transcriptase (TaKaRa RNA PCR kit, Takara,
| 14 | 16-01\00000000 |
Japan) in a conventional thermocycler. Specific gene amplification was performed using hot start TaKaRa Ex Taq DNA polymerase. Specific primers that do span intronic sequences were designed for mouse mRNA of eotaxin-2 and TGF-beta (Table 1). The following PCR conditions were used for amplification of G3PDH and TGF-beta: incubation of the samples at 95 °C for 2min and then 27 cycles (for G3PDH) or 35 cycles (for TGFbeta) consisting of 95 °C for 30 s; 55 °C for 45 s and 72°C for 1min. Touchdown PCR amplification protocol was used for analysis of eotaxin-2 mRNA expression: with the starting temperature 70°C and amplification for 30 cycles at annealing temperature of 60°C. PCR samples were run on a 2% agarose gel stained with Gelstar Nucleic Acid Stain (Gambrex, USA), and the PCR products were visualized with 300 nm UV transilluminator and photographed with Polaroid camera system. The absence of genomic contamination of the isolated RNA was confirmed by performing glyceraldehyde-3-phosphate dehydrogenase (G3PDH) PCR reactions on the purified RNA samples prior to reverse transcription. A negative control of nuclease-free water was included with all sample runs. Each RNA sample was analyzed several times.
Immunohistochemical Analysis of aortic sinus
Cryostat sections (5 µm thick) of the aortic sinus were evaluated employing indirect immunoperoxidase staining. Slides were than counterstained with Xxxxx’x hematoxylin and mounted with glycerol (Dako). Immunohistochemical staining was performed employing affinity purified goat anti-murine eotaxin-2 (Cytolab, Israel).
Statistical analysis
Comparison between groups was done by the one-way Anova test. P<0.05 was considered statistically significant. Results are expressed as means and standard error.
| 15 | 16-01\01840495 |
Examples
Example 1: Specificity and Anti-inflammatory effect of an anti eotaxin-2 mAb (D8)
Several clones of Anti eotaxin-2 murine monoclonal antibodies (mAbs) were prepared as described above in the Materials and Methods section.
The specificity of the novel anti eotaxin-2 antibody was assessed in a binding assay. Serial dilutions of the mAbs produced in clone D8 were added to plates coated with either eotaxin or eotaxin-2, for an overnight incubation. After washing, the plates were incubated with a goat anti-mouse peroxidase conjugated antibody, washed again and binding was detected using a colorimetric substrate.
As shown in Figure 1, mAbs of clone D8 showed significant binding to eotaxin-2 and no binding to eotaxin, in all the range of tested dilutions.
The antibodies were also tested for their capability to inhibit adhesion of murine or rat splenocytes as well as human PBMCs to fibronectin or to attenuate their migration towards VEGF. D8 (50 µg) was found to inhibit adhesion of murine and rat splenocytes as well as human PBMC to fibronectin (FN) by 35-55% (Figure 2A). Migration towards VEGF was also attenuated in a dose-dependant manner (Figure 2B), confirming the potential anti-inflammatory effects of the antibody.
Example 2: Anti-inflammatory potential of clone D8 mAb (in vivo data)
A. Rheumatoid Arthritis model
Adjuvant arthritis was induced in Xxxxx rats (six-week-old male Xxxxx rats obtained from Xxxxxx, Xxxxxx) by injection of incomplete Xxxxxx’x adjuvant. Xxxxxx’x incomplete adjuvant was prepared by suspending heat-killed Mycobacterium
| 16 | 16-01\01840495 |
Tuberculosis (Difco, Detroit, MI) in mineral oil at 10 mg/ml. Rats were injected intradermally with 100 µl adjuvant at the base of the tail. Arthritis developed by day 10 post injection.
Evaluation of the effect of anti – eotaxin-2 antibodies, compared with nonspecific IgG and PBS as controls, on adjuvant induced arthritis:
Rats (8 per group) were treated by intraperitoneal (IP) injection with 3 monoclonal antibodies against Xxxxxxx-0, (X0, X0, D8) 3X/week. Controls were treated with total mouse (non specific) IgG or PBS. Injections were started on the third day after adjuvant administration and were performed three times a week until the rats were sacrificed.
Dose response experiments:
In a second set of experiments, D8, the anti-eotaxin-2 antibody showing best protective results in the adjuvant- induced arthritis model, was tested in a dose – response model. Adjuvant arthritis was induced according to the above described protocol. Animals (6 rats per arm) were treated with D8 intraperitonealy at a dose of 20µg, 100µg or 1000 µg, starting on day 3 after adjuvant injection, three times weekly (D8 prevention group). A separate set of animals (6 in each group) were treated with identical doses after arthritis onset, namely when arthritis was already evident (D8 treatment group).
In order to compare the anti-inflammatory efficacy of D8 with that of a traditional anti-inflammatory agent of known efficacy, one group was treated with methotrexate (ip), 0.25mg/kg, once weekly, starting on day 3 after adjuvant injection (methotrexate prevention group). An additional group was treated with methotrexate, 0.25 mg/kg once weekly, in combination with D8, 100 µg (ip injection) given 3 times a week, starting on day 3 (combined D8 – methotrexate prevention group). A control group was treated with PBS throughout the experiment.
| 17 | 16-01\01840495 |
Evaluation of arthritis severity:
Arthritis severity was evaluated by measuring body weight, paw swelling, arthritic score and whole animal mobility. Sample joints were obtained for pathological evaluation, and post mortem X-ray of ankle joints was performed to document erosions.
Body weight in grams was measured every other day as an indicator of systemic inflammation.
Evaluation of Paw swelling: Ankle and wrist diameter in mm (to one place after the decimal point) were recorded three times a week using a caliper.
Arthritic score measurement: Each paw was scored on a scale of 0-4 for the degree of swelling, erythema, and deformity (maximum score 16 per animal accounting for all four paws) as follows: 0= normal; 1=slight erythema and/or swelling of the ankle or wrist; 2=moderate erythema and/or swelling of ankle or wrist; 3=severe erythema and/or swelling of ankle or wrist; 4=complete erythema and swelling of toes or fingers and ankle or wrist, and inability to bend the ankle or wrist.
Finger and toe swelling was recorded according to their partial contribution: Ankles (feet): each toe scored 0.2; Wrist: each finger scored 0.25 Sum of all joints was calculated.
| 18 | 16-01\01840495 |
Mobility score:
Whole animal Mobility was scored between 0-4, according to the following definitions:
0= normal; 1= slightly impaired; 2= major impairment; 3= does not step on paw; 4= no movement.
Statistical analysis: QuickCalcs software (Graph-Pad Software, San Diego, CA) was used for statistical analysis. Student’s t-test was performed to identify significant differences between experimental groups.
RESULTS:
A. arthritis model
Significant inhibition of arthritis was observed in rats treated with the anti—eotaxin—2 antibodies, compared to those treated with immunoglobulin (IgG) or PBS. As demonstrated in Figures 3A-3C, inhibition by the antibodies was manifested in all the tested parameters (arthritic score, mobility score, and ankle diameter). The antibody marked D8 showed the most significant effect. In the the arthritic score test (Fig 3A) statistically significant differences (P < 0.05) were obtained at every measurement, from day 13 to day 21, when comparing rats treated with D8 to rats treated with PBS or to rats treated with IgG. The protective effect became evident immediately with appearance of arthritis, on day 17 after induction. It continued to increase in magnitude until the end of the experiment, on day 21.
In the mobility score test statistically significant differences (P < 0.05) were obtained at every measurement, from day 17, when comparing rats treated with D8 to rats treated with PBS, and from day 18 when comparing rats treated with D8 to rats treated with PBS or to rats treated with IgG. Thus, the average mobility score of animals treated with D8 was 1.37 on day 21 compared with 2.43 in animals treated with PBS (p=0.05).
| 19 | 16-01\01840495 |
In the ankle diameter tests statistically significant difference (p<0.05) was obtained between D8 and PBS as of day 19. The difference between D8 and IgG did not reach statistical significance.
As indicated above, D8 treated rats had lower scores of arthritis which ranged from 2.6 to 3.0 than rats treated with PBS (Fig. 4A). For the histological scoring, each paw was scored on a scale of 0-4 for the degree of destruction: 0= normal; 1= inflammatory infiltrates and synovial hyperplasia; 2= pannus formation and cartilage erosion; 3= important cartilage erosion and bone destruction; 4= loss of joint integrity. Histological analysis of the joints of the arthritic rats revealed that joints of D8 treated rats had synovial hyperplasia and scattered inflammatory infiltrates (Fig. 4B), while most of the rats treated with PBS (control group) had severe synovitis with xxxxx formation and an intense inflammatory infiltrate (Fig. 4C).
In order to evaluate the effect of treatment with anti-eotaxin-2 antibodies on the systemic inflammatory response, average weight of animals was documented. As shown in figure 5, anti-eotaxin-2 treatment significantly ameliorated the loss of weight caused by the systemic inflammatory response induced by adjuvant arthritis. Again, the maximal protective effect was observed in animals treated with the D8 antibody, which continued to gain weight throughout the experiment. Statistically significant difference in weight (p<0.05) between D8 and PBS was obtained on day 17.
| 20 | 16-01\01840495 |
Dose response experiments:
D8 prevention of arthritis:
In the series of dose response experiments, D8 at a dose of 100 µg had a significantly superior protective effect, compared with the low dose (20µg) and high dose (1000µg) groups (Figure 6A). Similar results were obtained regarding the mobility scores, ankle diameter and animal weight (data not shown). Statistically significant differences (P < 0.05) were obtained at every determination, from day 17 to day 24 when comapring rats treated with D8 100µ to rats treated with PBS.
D8 treatment of arthritis:
Treatment with D8 antibody intraperitonealy beginning at the time of appearance of arthritis also resulted in a significant reduction in arthritic score severity (Figure 6B) compared with PBS treated animals. Similar results were obtained regarding mobility, weight and ankle diameter. As demonstrated in Figure 6B, in this experimental set up similar results were obtained at the 100 µg and 1000 µg dose groups.
Combined D8 – methotrexate (MTX) prevention of arthritis:
While both methotrexate and D8 100 µg produced significant, comparable protection against development of arthritis, as measured by the arthritic score (compared to PBS treated controls), the combination of methotrexate and D8 produced an enhanced (synergistic) protective effect, as demonstrated in Figure 7.
Both D8 100 µg and MTX treatment caused a statistically significant effect (p<0.05) compared with PBS as of day 13. By the end of the experiment, on day 24, a statistically significant difference in the arthritis score was observed between rats treated with MTX alone and rats treated with MTX + D8 100 µg. A significant difference was also observed on day 24 in the mean ankle width between these groups (not shown).
| 21 | 16-01\01840495 |
X-ray results:
Post – mortem X-ray demonstrated an intense degree of peri-articular soft tissue swelling in PBS treated rats, compared with minimal swelling in rats treated with D8 (Figure 8). In addition, control rats showed signs of decalcification and early erosion, which was not evident in the D8 treated animals. This is indicative of a significant reduction of inflammation in D8 treated animals.
Similar results were seen in x-rays of the forefeet (not shown).
B. Colitis model
In order to induce chronic colitis, ten-week old C57BL mice underwent three cycles of exposure to dextrane sulfate (DSS) in their drinking water for five days followed by 10-day intervals with regular tap water. By the end of the first cycle, mice were randomized into six treatment arms: vehicle control (PBS), total mouse IgG, and D8 given at increasing doses of 5µM, 25µM, 100µM and 200µM. Treatment was given by intraperitoneal (ip) injection in a 3X/week regimen. Body weight was documented twice a week. By the end of the last cycle, mice were sacrificed; the proximal portions of the colon were taken for immunohistochemical analysis and the distal portion for myeloperoxidase (MPO) activity assay (according to standard protocols) in order to assess the degree of the induced inflammation. The levels of inflammatory cytokines in the animals’ sera were detected by flow cytometry.
Treatment with IgG or with D8 significantly attenuated body weight loss compared to vehicle-treated animals. Throughout the study, the highest body weight was observed in the 5µg D8 treatment arm (Figure 9A). MPO activity on day 34 was significantly reduced in the 5µg D8 treatment group compared to all other treatment arms including mIgG (Figure 9B). In addition, the level of inflammatory cytokines in the sera of the D8-treated animals was lower than that detected in control animals (Figure 9C). Immunohistochemical analysis of the proximal colon confirmed reduction in the level of damage to the colon tissue as well as reduction in the extent and degree of inflammation (Figure 9D). The extent of inflammatory infiltrate was evaluated by an expert pathologist.
| 22 | 16-01\01840495 |
C. EAE
EAE serves as a typical animal model to study potential therapeutics for the human disease multiple sclerosis (30).
Ten-week old C57BL mice were injected subcutaneously (sc) with 200µg MOG (Myelin Oligodendrocyte Glycoprotein) peptide suspended in Complete Xxxxxx’x adjuvant, followed by a second injection one week later. One day after induction of the disease (namely, one day after the second injection) treatment with vehicle control (PBS), total mouse IgG, 25µg D8 and 100µg D8 (3X/week, ip injections) commenced. The severity and the progression of the disease was documented 3X/week according to the standard EAE scoring system (31).
Treatment with 25µg, and more significantly with 100µg of D8, attenuated the progression of EAE signs during the whole course of the experiment. Moreover, D8 at the higher dose (100 µg) reduced the incidence of the disease from about 90% (in all other treatment groups) to only 55% (Figure 10), thus shedding light on the potential therapeutic advantage of D8 in the treatment of EAE.
D. Diabetes
The non-obese diabetic (NOD) mouse serves as an animal model of autoimmune diabetes (32).
Six-week old NOD mice were treated 3X/week with D8 or with vehicle control (PBS). Between days 49 to 112, the disease incidence was tested using a commercial urine test and the eotaxin-2 levels in their sera on day 112 was determined using an ELISA (Mouse CCL24/Eotaxin-2/MPIF-2 DuoSet; R&D).
| 23 | 16-01\01840495 |
Diabetic incidence was markedly reduced in the D8-treated group compared to the untreated control (Figure 11A). In line with the clinical improvement, the level of eotaxin-2 was greatly reduced in the sera of the anti-eotaxin-2-treated animals compared to the control ones (Figure 11B).
E. Inhibition of atherosclerotic plaque formation
The expression level of an array of inflammatory cytokines and chemokines was measured in atherosclerotic lesions (plaques). Vulnerable plaques recovered from culprit coronary arteries of patients with acute myocardial infarction were compared with stable plaques obtained from endarterectomy samples. An analysis of the plaques by protein arrays is shown in Fig. 12. Processing was done as described in the materials and methods section.
Among the differentially expressed proteins, a significant alteration was found in the following proteins: XXXX-0, Xxxxxxx-0, XX-00, MCP-1 and TIMP-2; all exhibited a more than twofold reduction in expression in vulnerable versus stable plaques (Fig12A).
In immunohistochemistry studies in atherosclerosis prone mice (apoE KO mice), fatty streaks and advanced lesions from young and older mice were stained with anti-eotaxin-2 abs as described in materials and methods. Eotaxin-2 was shown to be present within endothelial cells and within plaque macrophages (Fig. 13).
mRNA expression was measured in young (6 week old) and atherosclerotic apoE KO mice. Aortas were obtained from the mice and subjected to RT-PCR as described in materials and methods. mRNA levels of eotaxin-2 and TGF-beta were assayed comparatively. Eotaxin-2 mRNA levels were found to be significantly higher in the young versus the older mice and this expression pattern paralleled the one observed with regard to the anti-atherosclerotic agent TGF-beta. Fig. 14A shows representative examples from each group.
| 24 | 16-01\01840495 |
Oxidized LDL is considered to play a key role in promoting atherogenesis. Mouse H5V endothelial cells were incubated with oxidized LDL (oxLDL) (1µg/ml). oxLDL significantly upregulated eotaxin-2 mRNA levels in murine H5V endothelial cells (Fig. 14B).
To determine whether eotaxin-2 has a role in the adhesion of cellular components of plaque inflammation, adhesion assays were performed on cultured endothelial cells. Splenocytes from either young or older atherosclerotic apoE KO mice were isolated from the spleen. Murine endothelial cells were incubated with oxLDL (1µg/ml) and the adhesion of the splenocytes onto the endothelial cells was examined in the presence of eotaxin 2 or control IgG antibodies (Fig 15A). Preincubation of the endothelial cells with blocking antibodies to eotaxin-2 was found to attenuate the adhesion of the splenocytes to these cells (Fig. 15A). This effect was more robust in lymphocytes from atherosclerotic (6 months old) apoE KO mice compared to those obtained from young non-atherosclerotic mice (aged 6 weeks). These findings were also evident when monocyte-macrophage cell line (U937 cells) was allowed to adhere to the cultured endothelial cells (H5V murine endothelial cell line) (Fig. 15B).
The effect of eotaxin-2 blockade on early and advanced atherosclerotic plaques was measured. In preliminary studies, it was found that administration of twice weekly doses of 5 µg of blocking anti-eotaxin-2 antibodies were sufficient to significantly reduce eotaxin-2 mRNA levels in the aortas of the mice. Next, the effect of short term administration of anti-eotaxin-2 abs was examined. Young apoE KO
| 25 | 16-01\01840495 |
mice were treated with Eoaxin-2 antibodies twice a week (i.p injections of 5 µg), or with control IgG or with PBS for 4 weeks and the effect on fatty streaks was measured. The mice were sacrificed for analysis of plaque size after oil-red O staining. Anti-eotaxin-2 drastically reduced fatty streak formation as compared to mouse IgG by approximately 72 percent (Fig. 16A). This effect was not associated with a change in lipid profile as total cholesterol and triglycerides were similar in both groups (data not shown). Moreover, treatment with control murine IgG did not influence plaque progression in comparison with PBS injections.
Next the effects of eotaxin-2 blockade were tested in a long-term model in which plaque architecture is more complex. Herein, after 10 weeks of two weekly injections of eotaxin-2 antibodies (5µg/dose), no significant differences were evident with regard to plaque size as measured in the hearts of older apoE KO mice (Fig. 16B). However, when plaque stability measured by fibrous area was assayed, it was found that antibodies to eotaxin-2 induced a significantly more stable plaque phenotype evident by a larger fibrous area at the expense of a smaller lipid core (Fig. 16C). Again, these findings were irrespective of lipid levels that were not different between groups. Representative Oil-red O and masson’s trichrome stained sections are provided in figure 17.
Atherosclerosis is a process in which fat deposition progresses in the arterial wall leading to progressive narrowing of the lumen. The mature plaque is composed of two basic structures: the lipid core and the fibrous cap. The smaller the lipid core and the thicker the fibrous cap, the more stable the plaque is, meaning that its propensity to rupture and cause myocardial infarction or unstable angina are increased. It is now clear that most plaques that cause acute coronary syndromes (e.g., myocardial infarction and unstable angina) are angiographically shown to have <70%
| 26 | 16-01\01840495 |
stenosis (reviewed in 28, 29). Approximately 60% of these lesions are caused by rupture of plaques with a large thrombogenic core of lipid and necrotic debris (including foci of macrophages, T cells, old hemorrhage, angiogenesis, and calcium). The ruptured cap is thin, presumably because macrophages secrete matrix metalloproteinases that digest it as they move across plaque, and because smooth muscle cells (the supporting element of the plaque) are depleted due to senescence or apoptosis caused by several factors, such as inflammatory cytokines.
In one of its aspects, the present invention is based on the finding that Eotaxin-2 is differentially expressed in stable versus vulnerable human atherosclerotic plaques. By blocking the Eotaxin-2 pathway in an apoE knockout (KO) [20] mouse model, the inventors were able to demonstrate both inhibition of fatty streak formation (which signifies early atherosclerotic lesions) and prolongation of plaque stabilization.
The inventors of the present invention found that eotaxin-2 is expressed in the endothelium of atherosclerotic and non atherosclerotic murine arteries supporting previous reports in the art. However, the inventors have also found that eotaxin-2 is expressed in plaque macrophages. Eotaxin-2 was more abundantly expressed in the aortas of young apoE KO mice as compared to atherosclerotic apoE KO mice. Suggesting that eotaxin-2 is involved in the initial steps of atherosclerosis that comprise cell to cell adhesion of monocytes/macrophages to the endothelium. Indeed, the in-vitro studies described below show that blocking eotaxin-2 reduces oxLDL induced adhesion of lymphocytes to endothelial cells, supporting a role for eotaxin-2 in plaque formation in vivo.
| 27 | 16-01\01840495 |
Despite intensive research, the factors that govern the transition of a stable to vulnerable plaque remain elusive. Based on an inflammatory protein array analysis of stable versus vulnerable human plaques, the present invention provides a potential target protein, eotaxin-2, to be involved in the transition of the plaque between a stable and a vulnerable phenotype. Whereas solid data exists with respect to the association of atherosclerosis with VCAM-1, IL-10 and MCP-1, which were also found by the inventors to be differentially expressed in stable versus vulnerable plaques, no such data exists for eotaxin-2.
In addition, eotaxin-2 was found by the inventors to be expressed in the endothelium of atherosclerotic and non atherosclerotic murine arteries thus supporting previous reports. However, it was also found by the inventors to be expressed in plaque macrophages. Interestingly, eotaxin-2 was more abundantly expressed in the aortas of young versus atherosclerotic apoE KO mice corresponding to initial steps of atherosclerosis that comprise cell to cell adhesion of monocytes/macrophages to the endothelium. Indeed, the in-vitro studies support a role for eotaxin-2 blockade in oxLDL mediated adhesion as may well occur in vivo. The more robust expression of eotaxin-2 in early stages of murine atherosclerosis also explains the impressive effect of blocking this pathway in the short term fatty streak model. Without wishing to be bound by theory, blocking inflammatory cell adhesion to the endothelium may be principally responsible for this effect.
With respect to the effects of eotaxin-2 blockade on plaque stability as evident by fibrous area, if inflammatory cell recruitment is attenuated due to eotaxin-2 blockade, it may be anticipated that the cytokine milieu will be more favorable towards a stable phenotype. These findings may not necessarily be reflected in a reduced extent of atherosclerosis as plaque built up is more likely to be influenced by lipid profile that has not changed due to eotaxin-2 blockade.
| 28 | 16-01\01840495 |
References
1. Xxxxx A LA. Chemokines and their receptors: drug targets in immunity and inflammation. Annu Rev Pharmacol Toxicol 2008;48:171-97.
2. Xxxx PJ, Xxxxxxxxx-Xxxxxxx DA, Xxxxxxx XX et al., Eotaxin: a potent eosinophil chemoattractant cytokine detected in a guinea pig model of allergic airways inflammation, J Exp Med 1994; 179: 881–887.
3. Kitaura M, Nakajima T, Imai T, et al., Molecular cloning of human eotaxin, an eosinophil-selective CC chemokine, and identification of a specific eosinophil eotaxin receptor, CC chemokine receptor 3, J Biol Chem 1996; 271: 7725–7730.
4 Xxxxxx XX, Qin S, Xxxxxxx XX, et al., Cloning of the human eosinophil chemoattractant, eotaxin. Expression, receptor binding, and functional properties suggest a mechanism for the selective recruitment of eosinophils. J Clin Invest 1996; 97: 604–612.
5. Xxxxxxxx V, Xxxxxxxxxx G, Xxxxxxxx XX, et al., Eotaxin and CCR3 are up–regulated in exacerbations of chronic bronchitis, Allergy 2002; 57: 17–22.
6. Xxxxxxxxx P, Xxxxxxxxxx F, Lasagni L, Xxxxxxx E, Beltrame C, Francalanci M,. Xxxxxxxxx M, Xxxxx G, Cosmi L, Maurenzig L, et al. Cell cycle-dependent expression of CXC chemokine receptor 3 by endothelial cells mediates angiostatic activity. J. Clin. Invest. 2001; 107:53.
| 29 | 16-01\01840495 |
7. Xxxxxx O, Gan X, Gujuluva C, Xxxxx AR, Sulur G, Stins M, Way D, Xxxxx M, Xxxxxxx M, Said J, Xxx KS, Xxxx D, Xxxxxx MC, Xxxxx M. CXC and CC chemokine receptors on coronary and brain endothelia. Mol Med. 1999; 5: 795-805.
8. Cheng SS, Lukacs NW, Xxxxxx XX. Eotaxin/CCL11 suppresses IL-8/CXCL8 secretion from human dermal microvascular endothelial cells, J Immunol 2002; 168 2887–2894.
9. Xxxxxxx, R., X. X. Xxxxx, X. Xxxxxxxxx, X. X. Xxxxxx, X. Xxxxxxx, X. Xxxxxx, X. Xxxxxxxxx, X. X. Xxxxxxxxx. 2000. Differential expression and responsiveness of chemokine receptors (CXCR1–3) by human microvascular endothelial cells and umbilical vein endothelial cells. FASEB J. 14:2055.
10. Xxxxxxx, R., X. X. Xxxxx, X. X. Xxxxx, X. X. Xxxx, X. X. Xxxxxxxx, X. X. Xxxxxx, X. X. Xxxxxxxxx. 2001. Eotaxin (CCL11) induces in vivo angiogenic responses by human CCR3+ endothelial cells. J. Immunol. 166:7571
11. X.X. Xxxxxx, X.X. Xxx and I.I. Galaria et al., CCL11 (eotaxin) induces CCR3-dependent smooth muscle cell migration, Arterioscler Thromb Vasc Biol 24 (2004), pp. 1211–1216.
12. Forssmann U. et al. Eotaxin-2, a novel CC chemokine that is selective for the chemokine receptor CCR3, and acts like eotaxin on human eosinophil and basophil leukocytes X.Xxx.Xxx. 1997; 185:2171-2176
13. Xxxxxxxxx P AF, Lasagni L, Xxxxxxx E, Xxxxxxxx C, Francalanci M, Xxxxxxxxx M, Xxxxx G, Cosmi L, Maurenzig L, Baggiolini M, Maggi E, Xxxxxxxxx S, Xxxxx M. Cell cycle-dependent expression of CXC chemokine receptor 3 by endothelial cells mediates angiostatic activity. J Clin Invest 2001;107(1):53-63.
| 30 | 16-01\01840495 |
| 14. | Xxxxxx G GV, Humbert M. New chemokine targets for asthma therapy. Curr Allergy Asthma Rep 2005;5(2):155-60. |
15. Xxxxxxxxx XX VR, Xxxxx XX, Xxxxxxx MB. Chemokines and chemokine receptors in mucosal homeostasis at the intestinal epithelial barrier in inflammatory bowel disease. Inflamm Bowel Dis 2008;14(7):1000-11.
16. Xxxxxxx J RP, Xxxxxxxx J, Xxxxxx XX, Male D, Woodroofe MN. Expression of the beta-chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J Neuroimmunol 2000;108(1-2):192-200.
| 17. | Xxxxxxxxx XX. Evolving concepts of rheumatoid arthritis. Nature 2003;423(6937):356-6. |
| 18. | Xxxxx XX. Physiology of cytokine pathways in rheumatoid arthritis. Arthritis Rheum 2001;45(1):101-6. |
19. Guglielmotti A XXX, Xxxxxxx I, Aquilini L, Milanese C, Pinza M. Amelioration of rat adjuvant arthritis by therapeutic treatment with bindarit, an inhibitor of MCP-1 and TNF-alpha production. Inflamm Res 2002;51(5):252-8.
20. Xxxx XX MR, Xxxxx N, Xxxxxx XX 3rd, Xxxxxxxx XX, Xxxx XX. Chemokine receptor expression in rat adjuvant-induced arthritis. Arthritis Rheum 2005;52(12):3718-30.
21. Xxxxxxxxx XX ST, Xxxxxxxx-Xxxxxxxx P, Tak PP. Chemokine and chemokine receptor expression in paired peripheral blood mononuclear cells and synovial tissue of patients with rheumatoid arthritis, osteoarthritis, and reactive arthritis. Xxx Rheum Dis 2006;65(3):294-300.
| 22. | Charo IF, Taubman MB. Chemokines in the pathogenesis of vascular disease. Circ Res. 2004; 95: 858-66. |
| 31 | 16-01\01840495 |
23. Sheikine S, Xxxxxxx XX. Chemokines as potential therapeutic targets in atherosclerosis, Curr Drug Targets 2006; 7: 13–28.
24. Xxxxx XX, Xxxxx XX, Xxxx XX, et al., Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: using genomic technology to identify a potential novel pathway of vascular inflammation, Circulation 2000; 102: 2185–2189.
25. Sheikine Y, Xxxxx B, Gharizadeh B, Jatta K, Tornvall P, Ghaderi M. Influence of eotaxin 67G>A polymorphism on plasma eotaxin concentrations in myocardial infarction survivors and healthy controls. Atherosclerosis. 2006; 189: 458-63.
26. Emanuele E, Xxxxxxx C, X’Xxxxxx A, Xxxxxxxxx P, Xxxxx XX, Bertona M, Geroldi D. Association of plasma eotaxin levels with the presence and extent of angiographic coronary artery disease. Atherosclerosis. 2006 May;186(1):140-5.
27. X. Xxxxxxxx, X. Xxxxxxxxx and A. Katinioti et al., Chemokines in patients with ischaemic heart disease and the effect of coronary angioplasty, Int J Cardiol 80 (2001), pp. 55–60.
28. Naghavi M, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circulation. 2003; 108: 1664-72.
29. Naghavi M, et al. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part II. Circulation. 2003; 108: 1772-8.
| 30. | Mendel I et al. Eur J Immunol 1995 25:1951-9 |
| 31. | Xxxxxxx M et al. Neurosci Lett. 2007. 412: 24-8 |
| 32. | Xxxxxxxxx XX, and Singh B. Immunity 1997 Dec; 7(6):727-38. |
| 32 | 16-01\01840495 |
00. Xxxxxx X, Xxxx X, Xxxxxxx X, Xxxx-Xxxx A Shaish A, Levkovitz H, Blank M, Harats D, Shoenfeld Y. Induction of early atherosclerosis in LDL receptor deficient mice immunized with beta 2 glycoproein I. Circulation. 1998; 15: 1108-1115.
Table 1
| mRNA target |
primers |
Product size |
Accession no. | |||
| Eotaxin-2 | CTGTGCCTGACCTCCAGAAC
CTAAACCTCGGTGCTATTGC |
379bp | AF281075 | |||
| TGFbeta | CTTGGGCTTGCGACCCACGTAGTA
AGACGGAATACAGGGCTTTCGATTCA |
492bp | NM_011577 | |||
| G3PDH | AGCCCATCACCATCTTCCAG
CCTGCTTCACCACCTTCTTG |
585bp | NM_199472 | |||
| 33 | 16-01\01840495 |
CLAIMS:
| 1. | A pharmaceutical composition for treating inflammatory or autoimmune diseases comprising an Eotaxin-2 antagonist and a pharmaceutically acceptable carrier or excipient. |
| 2. | A pharmaceutical composition according to claim 1 wherein the Eotaxin-2 antagonist is an anti Eotaxin-2 antibody. |
| 3. | A pharmaceutical composition according to claim 2 wherein the anti-eotaxin-2 antibody is a monoclonal antibody or a polyclonal antibody. |
| 4. | A pharmaceutical composition according to claim 2 or 3 wherein said antibodies are human antibodies. |
| 5. | A pharmaceutical composition according to claim 2 wherein said antibody is a humanized antibody or a chimeric antibody. |
| 6. | A pharmaceutical composition according to claim 2 wherein said antibody is a monoclonal antibody secreted by hybridoma D8. |
| 7. | A pharmaceutical composition according to claim 1 wherein the Eotaxin-2 antagonist is an antisense or a siRNA molecule directed against Eotaxin-2 mRNA. |
| 8. | A pharmaceutical composition according to claim 1 wherein the Eotaxin-2 antagonist is a small molecule. |
| 9. | A method for treating inflammatory or autoimmune diseases comprising administering to a patient in need thereof an eotaxin-2 antagonist or a pharmaceutical composition according to any of claims 1-8. |
| 10. | A method according to claim 9 wherein said autoimmune disease is selected from the group consisting of rheumatoid arthritis, inflammatory bowel disease, colitis, and diabetes. |
| 34 | 16-01\01840495 |
| 11. | A method for inhibiting atherosclerotic plaque formation comprising administering to a patient in need thereof an Eotaxin-2 antagonist or a pharmaceutical composition according to any of claims 1-8. |
| 12. | A method for stabilizing an atherosclerotic plaque comprising administering to a patient in need thereof an eotaxin-2 antagonist or a pharmaceutical composition according to any of claims 1-8. |
| 13. | A method for preventing major cardiovascular events in a patient with acute coronary syndrome comprising administering to said patient an eotaxin-2 antagonist or a pharmaceutical composition according to any of claims 1-8. |
| 14. | A method according to any of claims 9-13 wherein said eotaxin-2 antagonist or said pharmaceutical composition according to any of claims 1-8 is administered in combination with at least one additional therapeutic agent. |
| 15. | A method according to claim 14 wherein said at least one additional therapeutic agent is selected from a group consisting of chemotherapeutics, cytokines, peptides, antibodies and antibiotics. |
| 16. | A method according to claim 15 wherein said at least one additional therapeutic agent is selected from the group consisting of cyclosporine, a steroid, a NSAIDS (non-steroidal anti inflammatory drug), anti-TNFa antibodies, anti-IL6 R antibodies, anti-CD20 antibodies, anti- VLA-4 antibodies IVIG, copaxone, and interferon-ß. |
| 17. | A method according to claim 15 wherein said at least one additional therapeutic agent is methotrexate. |
| 18. | A method according to claim 14 wherein said at least one additional therapeutic agent is administered simultaneously with said eotaxin-2 antagonist or said pharmaceutical composition. |
| 35 | 16-01\01840495 |
| 19. | A method according to claim 14 wherein said at least one additional therapeutic agent and said eotaxin-2 antagonist or said pharmaceutical composition are administered sequentially. |
| 20. | A hybridoma cell line secreting an anti-eotaxin 2 monoclonal antibody, wherein said hybridoma is D8, G7, or G8. |
| 21. | A monoclonal antibody directed against eotaxin-2, or any fragment thereof which retains the binding ability to eotaxin 2, wherein said monoclonal antibody is secreted from hybridoma D8, G7 or G8. |
| 36 | 16-01\01840495 |
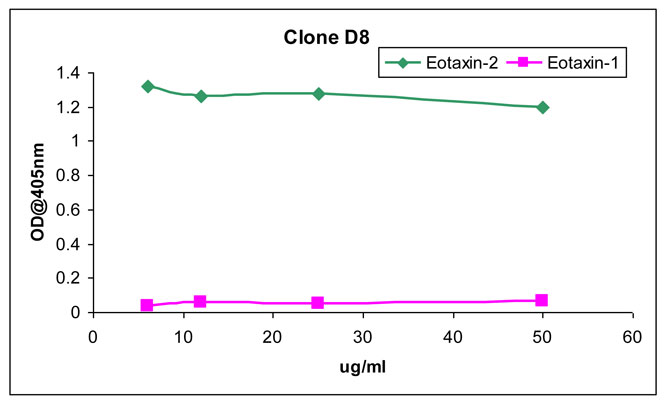
Fig. 1
| 37 | 16-01\01840495 |
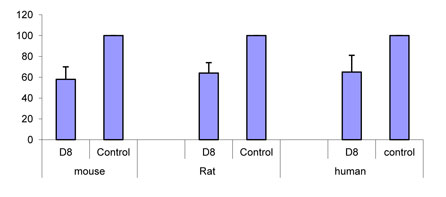
Fig. 2A
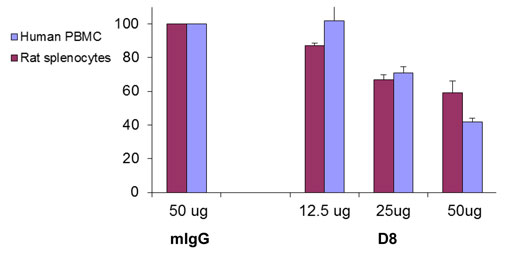
Fig. 2B
| 38 | 16-01\01840495 |
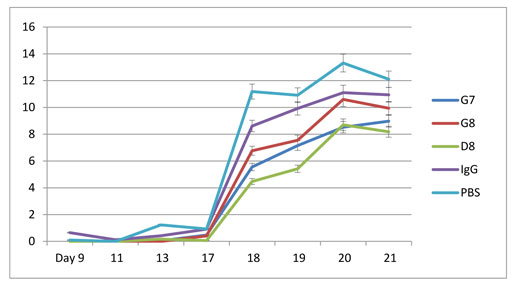
Fig. 3A
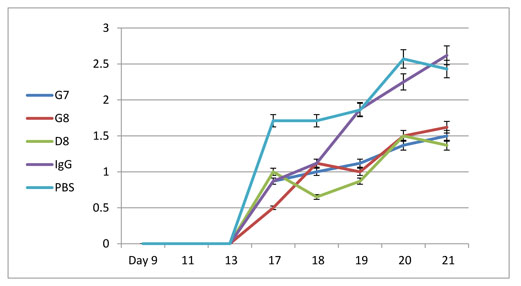
Fig. 3B
| 39 | 16-01\01840495 |
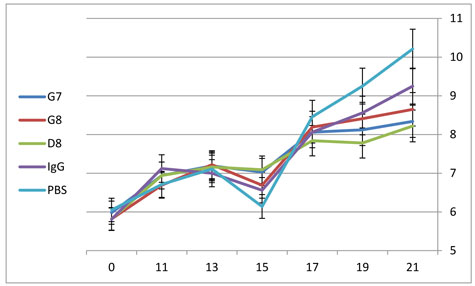
Fig. 3C
| 40 | 16-01\01840495 |
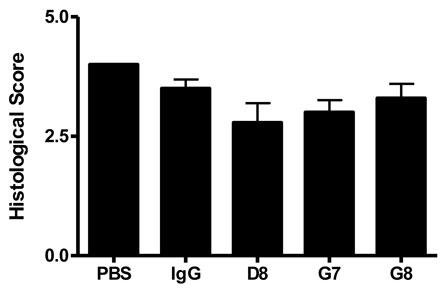
Fig. 4A
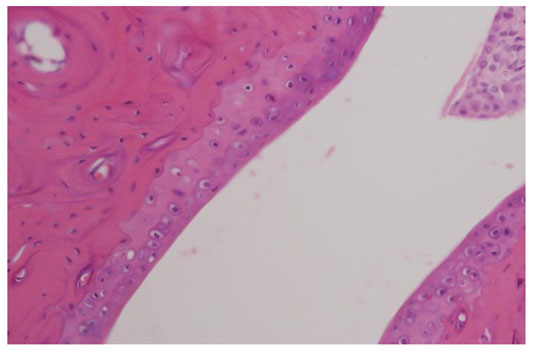
Fig. 4B
| 41 | 16-01\01840495 |
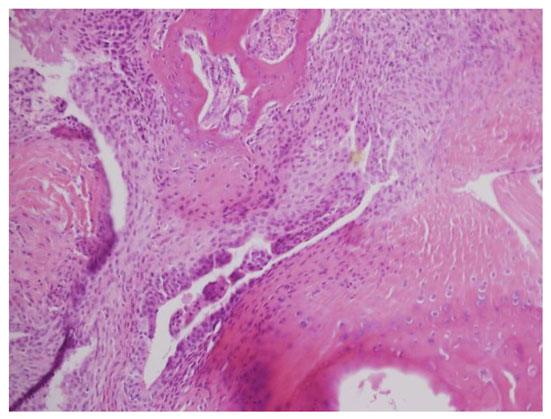
Fig. 4C
| 42 | 16-01\01840495 |
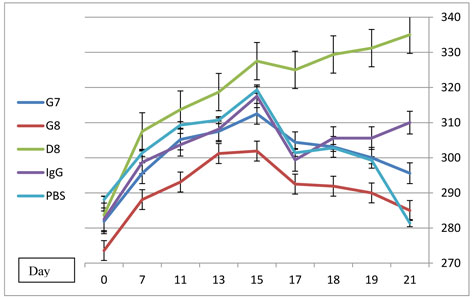
Fig. 5
| 43 | 16-01\01840495 |
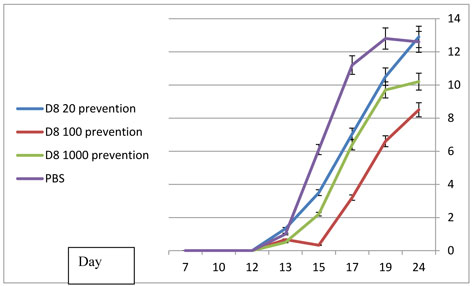
Fig. 6A
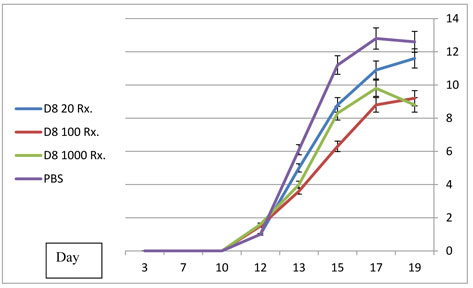
Fig. 6B
| 44 | 16-01\01840495 |
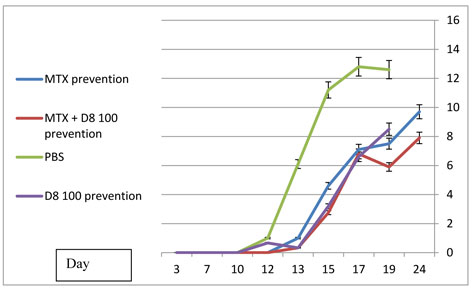
Fig 11B
| 45 | 16-01\01840495 |
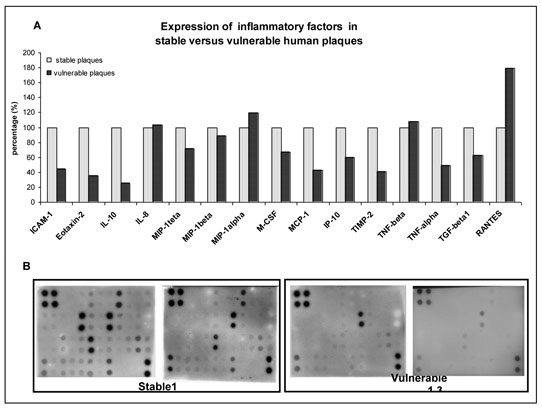
Fig 12
| 46 | 16-01\01840495 |
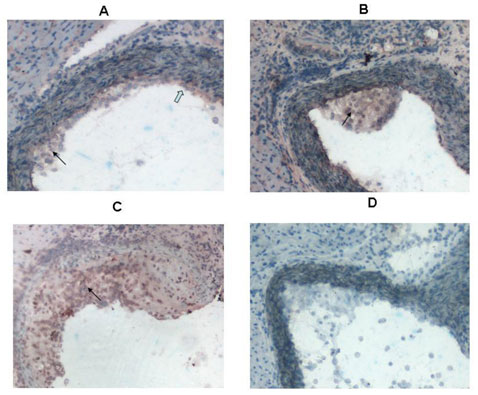
Fig 13
| 47 | 16-01\01840495 |
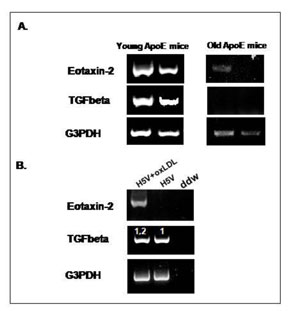
Fig 14
| 48 | 16-01\01840495 |
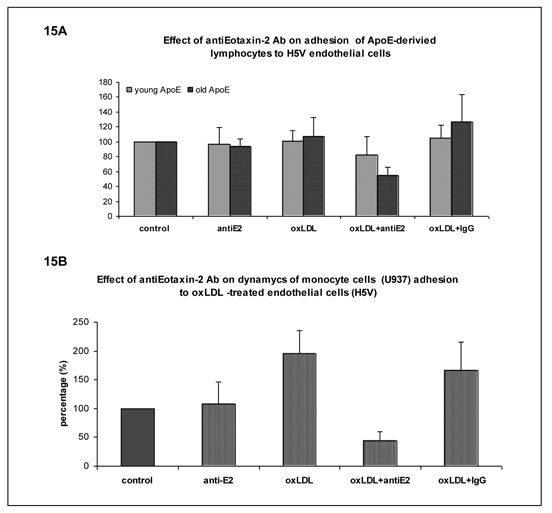
Fig 15
| 49 | 16-01\01840495 |
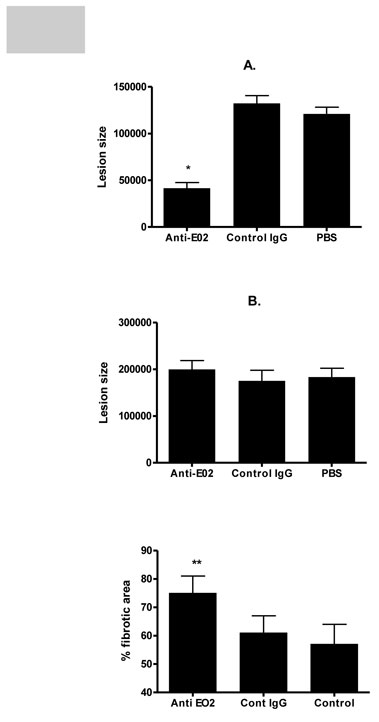
Fig 16
| 50 | 16-01\01840495 |
Fig 17
| 51 | 16-01\01840495 |
AMENDMENT TO AGREEMENT
THIS AMENDMENT TO AGREEMENT (this “Amendment”) is made and entered into on August 1, 2012, by and between Vascular Biogenics Ltd., an company registered under the laws of the State of Israel (“VBL”) and Xxxx. Xxxxx Xxxxxx (“Xxxxxx”). VBL and Xxxxxx shall be sometimes referred to each as a “Party” and collectively as the “Parties”).
W I T N E S S E T H:
WHEREAS, the Parties entered into a certain Agreement dated January 24, 2010 (the “Agreement”); and
WHEREAS, the Parties wish to amend the Agreement according to the terms hereof;
| 1. | Definitions; Effectiveness of Amended Agreement |
| 1.1. | Capitalized terms used herein and not otherwise defined shall have the respective meaning ascribed to them in the Agreement. |
| 1.2. | Other than as specifically amended hereby, the provisions of the Agreement shall remain in full force and effect. In the event of contradiction between any provision of the Agreement and an amendment thereto as set forth herein, such amendment shall prevail. |
| 2. | Amendment |
Revenue Sharing. Effective as of August 1, 2012, Section 2 of the Agreement shall be deleted in its entirety and replaced with the following:
In the event that Xxxxxx, or anyone in his behalf, receives from Sourasky Royalties, defined below, in connection with and/or as a result from the commercialization or otherwise exploitation of the Other IP or any other intellectual property if it is later found to be owned by Sourasky due to the engagement of Xxxxxx therewith, regardless of the Parties agreement in Section 1 above, then Xxxxxx undertakes to transfer or ensure the receipt by VBL of 50% of the Royalties within 30 days following the receipt thereof. In the event that the transfer of such portion of the Royalties is not feasible, the Parties shall agree on an appropriate mechanism to ensure VBL rights therein.
“Royalties” shall mean any proceeds or proprietary interest resulting from the commercial exploitation of any invention whether in cash or in kind.
1
[Signature Page to Amendment to Agreement]
IN WITNESS WHEREOF, the parties hereto have executed this Amendment to Agreement as of the date first written above.
| /s/ Xxxx Xxxxxx | /s/ Xxxxx Xxxxxx | |||
| VASCULAR BIOGENICS LTD. | XXXX. XXXXX XXXXXX |
| By: | Xxxx Xxxxxx | |
| Title: | CEO | |

