COLLABORATION AND LICENSE AGREEMENT
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
THIS COLLABORATION AND LICENSE AGREEMENT (“Agreement”) is entered into as of September 3, 2019 (the “Effective Date”), by and between MUNDIPHARMA MEDICAL COMPANY, a general exempted partnership established and existing under the laws of Bermuda, and having its principal place of business at Xxx Xx Xxxxx Xxxxx, 00 Par La Ville Road, Xxxxxxxx HM08, Bermuda (“Mundipharma”), and CIDARA THERAPEUTICS, INC., a corporation organized under the laws of the State of Delaware, USA, having its principal offices at 0000 Xxxxx Xxxxx Xxxxx, Xxxxx 000, Xxx Xxxxx, Xxxxxxxxxx 00000, XXX (“Cidara”).
RECITALS
WHEREAS, Mundipharma is engaged in the research, development and commercialization of pharmaceutical products;
WHEREAS, Cidara is developing, and possesses intellectual property rights and other proprietary information related to, its proprietary drug candidate known as rezafungin; and
WHEREAS, Mundipharma desires to obtain, and Cidara is willing to grant to Mundipharma, (a) a license to develop, register and commercialize rezafungin in an intravenous formulation in the Field in the Mundipharma Territory, (b) a worldwide license to manufacture rezafungin in an intravenous formulation, and (c) an option to obtain a license to develop, register and commercialize rezafungin in a formulation for subcutaneous administration and other formulations in the Field in the Mundipharma Territory; in each case, on the terms and subject to the conditions set forth in this Agreement.
AGREEMENT
NOW, THEREFORE, in consideration of the foregoing premises and the mutual covenants contained herein and other good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, the parties agree as follows:
1. | DEFINITIONS |
1.1 “Acceptance for Filing” or “Accepted for Filing” shall mean, with respect to an MAA filed: (a) in a Major European Country, the receipt of written notice of acceptance for filing of such MAA from (i) the EMA or (ii) the applicable Regulatory Authority in the applicable Major European Country (as applicable); or (b) in China, the receipt of written notice of acceptance of filing of such MAA from the NMPA.
1.2 “Accounting Standards” shall mean (a) U.S. generally accepted accounting principles or (b) International Financial Reporting Standards; in each case, as applicable, consistently applied throughout the organization of a particular Entity and its Affiliates.
1.3 “Act” shall mean, as applicable, the United States Federal Food, Drug and Cosmetic Act, 21 U.S.C. §§301 et seq., and all related rules, regulations and guidelines, as any of the foregoing may be amended from time to time.
1.4 “Actual Combination Product Net Sales” shall have the meaning provided in Section 1.110.
1.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
1.5 “Affiliate” shall mean, with respect to any Entity (including a party to this Agreement), any other Entity controlled by, controlling, or under common control with such Entity; provided, however, that with respect to Mundipharma, “Affiliate” shall not include any Entity that is incorporated or otherwise legally established in the US or in Japan. For the purposes of this definition, the term “control” (including, with correlative meanings, the terms “controlled by” and “under common control with”) shall mean direct or indirect ownership, including ownership by one or more trusts with substantially the same beneficial interests, of 50% or more of the outstanding voting and equity rights of such Entity, or possession of the power to direct the management and policies of such Entity.
1.6 “Anti-Corruption Laws” shall mean the U.S. Foreign Corrupt Practices Act (15 U.S.C. §§78dd-1, et. seq.), as amended, the Organization for Economic Co-operation and Development (OECD) Convention on combating bribery of foreign public officials in international business transactions, the UK Xxxxxxx Xxx 0000, as amended, and any other applicable anti-corruption laws.
1.7 “Anti-Fungal Product” shall mean any anti-fungal product targeted for the treatment of fungal infections, but excluding any Product.
1.8 “Anti-Fungal ROFN” shall have the meaning provided in Section 4.6.
1.9 “Anti-Fungal ROFN Exercise Period” shall have the meaning provided in Section 4.6.
1.10 “Anti-Fungal ROFN Term” shall have the meaning provided in Section 4.6.
1.11 “Applicable Laws” shall mean the applicable provisions of any and all national, supranational, regional, state and local laws, treaties, statutes, rules, regulations, administrative codes, guidances, ordinances, judgments, decrees, directives, injunctions, orders, permits of or from any court, arbitrator, Regulatory Authority or governmental agency or authority having jurisdiction over or related to the subject item, including the Act, Anti-Corruption Laws, data privacy laws and Export Control Laws.
1.12 “Business Day” shall mean any day except a Saturday, Sunday or any other day on which commercial banks in New York, New York, U.S. are authorized or required by law to remain closed.
1.13 “Business Information” shall mean any confidential, proprietary, strategic or other information or data, regardless of whether it is in tangible form, including reports and information related to or regarding a party or its Affiliate’s business plans, business methodologies, strategies, specifications, customers, prospective customers, partners, suppliers, billing records, finance or capitalization, litigation matters, marketing information, forecasts, trade secrets, products or services.
1.14 “C.F.R.” shall mean the United States Code of Federal Regulations.
2.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
1.15 “China” shall mean the People’s Republic of China, excluding the special administrative regions of Hong Kong and Macau.
1.16 “Cidara” shall have the meaning first set out above.
1.17 “Cidara-Controlled Affiliate” shall mean any Entity that is controlled (as such term is defined in Section 1.5) by Cidara.
1.18 “Cidara General Manufacturing/Formulation Patents” shall mean any Cidara Patent in the Mundipharma Territory that claims inventions that are necessary or useful for the manufacture or formulation of both (a) Compound or Licensed Product and (b) any compound that is not a Compound or any product that is not a Licensed Product.
1.19 “Cidara Invention” shall mean any Invention made solely by one or more employees, consultants or contractors of Cidara and/or Cidara-Controlled Affiliates.
1.20 “Cidara Know-How” shall mean all Information and Data Controlled by Cidara and/or Cidara-Controlled Affiliates as of the Effective Date or during the Term that is necessary or useful for the development, registration, manufacture, use or commercialization of Compound or Licensed Product in the Field, including Cidara Inventions; but excluding: (a) any such Information or Cidara Inventions directed to manufacturing or formulation technology that is not actually used by or on behalf of Cidara or Cidara-Controlled Affiliates in the development, registration, manufacture, use or commercialization of Compound or Licensed Product; (b) Expanded Licensed Product Clinical Efficacy Data generated by or on behalf of Cidara or Cidara-Controlled Affiliates unless the parties have executed an Expanded Licensed Product Amendment with respect to the applicable Expanded Licensed Product; (c) Cidara Patents; and (d) Joint Technology.
1.21 “Cidara Loan and Security Agreement” shall mean the Loan and Security Agreement between Pacific Western Bank and Cidara dated October 3, 2016, as amended.
1.22 “Cidara Patents” shall mean all Patents Controlled by Cidara and/or Cidara-Controlled Affiliates as of the Effective Date or during the Term that claim inventions that are necessary or useful for (a) the development, registration, use or commercialization of Compound or Licensed Product in the Mundipharma Territory, or (b) the manufacture of Compound or Licensed Product worldwide; but, in each case, excluding: (i) any such Patents that claim manufacturing or formulation technology that is not actually used by or on behalf of Cidara or Cidara-Controlled Affiliates in the development, registration, manufacture, use or commercialization of Compound or Licensed Product; and (ii) the Joint Patents. The Cidara Patents as of the Effective Date are set forth on Exhibit A.
1.23 “Cidara Peak Incremental Net Sales Forecast” shall have the meaning provided in Section 4.3(a)(i).
1.24 “Cidara Product Marks” shall have the meaning provided in Section 8.8(a).
3.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
1.25 “Cidara Subcutaneous Peak Net Sales Forecast” shall have the meaning provided in Section 4.4(c)(ii).
1.26 “Cidara Technology” shall mean the Cidara Patents, Cidara Inventions and Cidara Know-How.
1.27 “Cidara Territory” shall mean: (a) the U.S.; and (b) Japan.
1.28 “CMC” shall mean chemistry, manufacturing and controls information and data required as part of an IND, NDA, MAA or other Product Filing.
1.29 “CMC Development Plan” shall have the meaning provided in Section 4.10(a).
1.30 “CMO” means a Third Party contract manufacturing organization.
1.31 “Combination Product” shall mean a Licensed Product comprising a fixed-dose combination of Compound and at least one Other Active in an intravenous formulation.
1.32 “Commercially Reasonable Efforts” shall mean, with respect to the efforts to be expended by a party with respect to any objective, the level of reasonable, diligent, good faith efforts that biotechnology or pharmaceutical companies typically devote to product candidates or products owned by them that are at a similar stage in their development or product life, taking into account efficacy, safety, anticipated and approved labeling, the competitiveness of alternative products in the marketplace, the patent, regulatory and other market exclusivity position of the product, the likelihood of regulatory approval (including pricing and reimbursement approval), the profitability of the product, pricing and reimbursement, and other relevant technical, legal, scientific and medical factors. As used in this Section 1.32, “biotechnology or pharmaceutical companies” shall mean companies in the biotechnology or pharmaceutical industry (as applicable) of a size and stage of development similar to that of such party, including having human pharmaceutical product candidates or products in a similar stage of development or product life to Compound or Licensed Product. Commercially Reasonable Efforts shall be determined on a country-by-country and Licensed Product-by-Licensed Product basis, and it is anticipated that the level of efforts constituting Commercially Reasonable Efforts in one country may differ from the level of efforts constituting Commercially Reasonable Efforts in another country, reflecting changes in the status of the Licensed Product and the country(ies) involved.
1.33 “Compound” shall mean: (a) rezafungin; (b) the Lead Compound; (c) […***…]; or (d) […***…].
1.34 “Confidential Information” shall have the meaning provided in Section 7.1.
1.35 “Control” or “Controlled by” shall mean, with respect to any Information, Patents or other intellectual property rights, possession by a party of the ability (whether by ownership, license or other right, other than pursuant to a license granted to such party under this Agreement) to grant access to, to grant use of, or to grant a license or a sublicense to, such Information, Patents
4.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
or other intellectual property rights without violating the terms of any agreement or other arrangement with any Third Party.
1.36 “Cost of Goods” shall mean, with respect to Licensed Product supplied by or on behalf of Cidara hereunder:
(a) in the case of Licensed Product manufactured by a Third Party, payments made to such Third Party for such Licensed Product, […***…], in each case, determined in accordance with Accounting Standards. […***…]; and
(b) in the case of Licensed Product manufactured by Cidara or its Affiliate, the actual, reasonable fully-allocated cost of manufacturing such Licensed Product (in accordance with GMP, if applicable), determined in accordance with Accounting Standards, consistently applied, and which will comprise […***…], in accordance with the normal accounting practices for all other products manufactured in the applicable facility.
As of the Effective Date, Cidara does not anticipate that Cidara or any of its Affiliates […***…]. However, […***…].
1.37 “Data” shall mean any and all results of research, preclinical and non-clinical studies, including in vitro, in vivo, and ex vivo studies, clinical trials and other testing of Compound or Licensed Product, and any and all other data generated by or on behalf of a party related to the development, manufacture or commercialization of Compound or Licensed Product, including biological, chemical, pharmacological, toxicological, safety, pharmacokinetic, clinical, CMC, analytical, quality control, mechanical, software, electronic and other data, results and descriptions.
1.38 “Disclosing Party” shall have the meaning provided in Section 7.1.
1.39 “Distributor” shall mean: (a) a Third Party distributor of Licensed Product that has no royalty or other payment obligations to Mundipharma or any of its Affiliates that are calculated based on amounts invoiced or received by such Third Party for sales of Licensed Product; or (b) a Third Party distributor of Licensed Product that (i) does not take title to Licensed Product, (ii) does not invoice Licensed Product sales to Third Party customers and (iii) is responsible only for inventory management and distribution with respect to Licensed Product on behalf of Mundipharma or its Affiliate.
1.40 “Effective Date” shall have the meaning first set out above.
1.41 “EMA” shall mean the European Medicines Agency or any successor agency thereto in the EU having substantially the same function.
1.42 “Entity” shall mean any corporation, general partnership, limited partnership, limited liability partnership, joint venture, estate, trust, company (including any limited liability company or joint stock company), firm or other enterprise, association, organization or entity.
1.43 “EU” shall mean the European Union or any successor union of European states thereto having a substantially similar function.
5.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
1.44 “Excluded Validation Batch” shall have the meaning provided in Section 1.59.
1.45 “Exclusive License” shall have the meaning provided in Section 2.1(a).
1.46 “Existing Patents” shall have the meaning provided in Section 9.2(a).
1.47 “Expanded Licensed Product” shall mean (A) a Licensed Product for one or more indications in the Field in addition to the Lead Indications (each, a “New Indication”), such as […***…]; (B) a Licensed Product containing the Lead Compound other than the Lead Indication Product to the extent not managed entirely through the change control procedure agreed pursuant to Section 4.10(d); (C) a Licensed Product containing any Compound other than the Lead Compound; or (D) a Combination Product. For clarity, Expanded Licensed Product excludes the Pediatric Licensed Product.
1.48 “Expanded Licensed Product Amendment” shall have the meaning provided in Section 4.3(a)(ii).
1.49 “Expanded Licensed Product Buy-In Fee” shall have the meaning provided in Section 4.3(c).
1.50 “Expanded Licensed Product Clinical Efficacy Data” shall mean clinical efficacy data generated in a clinical trial of an Expanded Licensed Product conducted by or on behalf of a party as permitted by (a) the first paragraph of Section 4.3(a) in the case of Cidara or (b) Section 4.3(b) in the case of an Independent Development Party.
1.51 “Expanded Licensed Product Global Development Plan” shall mean, on an Expanded Licensed Product-by-Expanded Licensed Product basis, a written plan for the conduct of Cidara-sponsored clinical trial(s), GLP Study(ies) and CMC development activities of an Expanded Licensed Product to be conducted by or on behalf of Cidara (which may include activities in or for the Mundipharma Territory that will be performed by Mundipharma and/or its Affiliates) in support of the clinical development of, and filing of XXXx for, such Expanded Licensed Product in the Major Markets.
1.52 “Expanded Licensed Product Mundipharma Territory Plan” shall mean, on an Expanded Licensed Product-by-Expanded Licensed Product basis, a written plan setting forth the Mundipharma Territory-Specific (mutatis mutandis) clinical trials and GLP Studies of an Expanded Licensed Product to be conducted by or on behalf of Mundipharma in the Mundipharma Territory that are required to support MAA filing and Regulatory Approval (excluding pricing and reimbursement approval) for such Expanded Licensed Product in the Mundipharma Territory but are not included in the Expanded Licensed Product Global Development Plan.
1.53 “Export Control Laws” shall mean: (a) all applicable U.S. laws and regulations relating to sanctions and embargoes imposed by U.S. Department of Treasury’s Office of Foreign Assets Control (or its successor office or other body having substantially the same function); (b) all applicable U.S. export control laws, including the Arms Export Controls Act (22 U.S.C. Ch. 39), the International Emergency Economic Powers Act (50 U.S.C. §§ 1701 et seq.), the Trading With
6.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
the Enemy Act (50 U.S.C. app. §§ 1 et seq.), the Export Administration Act of 1979 (50 U.S.C. app. §§ 2401 et seq.), International Boycott Provisions of Section 999 of the U.S. Internal Revenue Code of 1986, and all rules, regulations and executive orders relating to any of the foregoing, including but not limited to the International Traffic in Arms Regulations (22 C.F.R. §§ 120 et seq.), the Export Administration Regulations (15 C.F.R. §§ 730 et. seq.), and the regulations administered by the Office of Foreign Assets Controls of the United States Department of the Treasury; and (c) all export controls imposed on any Licensed Product by any country or organization or nation within the jurisdiction of which either party operates or does business.
1.54 “FDA” shall mean the United States Food and Drug Administration, or any successor agency thereto in the U.S.
1.55 “Field” shall mean all uses in humans and non‑human animals.
1.56 “First Commercial Sale” shall mean, with respect to a Licensed Product in a country, the first commercial transfer or disposition for value of such Licensed Product by a Selling Party to a Third Party in such country after the applicable Regulatory Authority of such country has granted Regulatory Approval of such Licensed Product for any indication in the Field (whichever indication is the first indication for which such Licensed Product receives Regulatory Approval in such country). For clarity, there shall only be one “First Commercial Sale” of a particular Licensed Product in a country, regardless of the number of indications for which the applicable Regulatory Authority of such country grants Regulatory Approval of such Licensed Product.
1.57 “GCP” shall mean current good clinical practices for the clinical development of a pharmaceutical products investigated in clinical trials as set forth in ICH E6 Guideline and implemented in applicable EU legislations.
1.58 “Generic Product” shall mean, with respect to Licensed Product that has received Regulatory Approval in a regulatory jurisdiction in the Mundipharma Territory and is being marketed and sold by Mundipharma or any of its Affiliates or Sublicensees in such jurisdiction, any pharmaceutical product that: (a) is sold in such jurisdiction by a Third Party that is not a Sublicensee of Mundipharma or its Affiliates and did not purchase or acquire such product in a chain of distribution that included Mundipharma or any of its Affiliates or Sublicensees; and (b) has received Regulatory Approval in such jurisdiction, for at least one of the same indications as such Licensed Product, as a “generic drug,” “generic medicinal product,” “bioequivalent” or similar designation of interchangeability by the applicable Regulatory Authority in such jurisdiction, pursuant to an expedited, abbreviated or bibliographic approval process in accordance with the then-current rules and regulations in such jurisdiction, where (i) such Licensed Product is the “reference medicinal product,” “reference listed product” or similar designation in such jurisdiction, and (ii) such approval referred to or relied on (x) the approved MAA for such Licensed Product held by Mundipharma, its Affiliate or a Sublicensee in such jurisdiction or (y) the data contained or incorporated by reference in such approved MAA for such Licensed Product in such jurisdiction.
1.59 “Global Development Costs” shall mean the reasonable and documented external costs and expenses incurred by Cidara and/or Cidara-Controlled Affiliates and, if applicable, by Mundipharma and/or its Affiliates, after the Effective Date in connection with (a) the performance
7.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
of Global Development Plan activities, or (b) preparing, applying for, obtaining or maintaining any IND or other Product Filings specific to any Global Development Plan activity (for clarity, excluding any NDA, MAA or Regulatory Approval). Specifically, Global Development Costs of a particular activity shall comprise the following: (i) all reasonable and documented amounts paid by a party to Third Party contractors and consultants and other Third Parties in connection with such Global Development Plan activity, including, without limitation, any such amounts paid to a Regulatory Authority to apply for, obtain or maintain any IND or other Product Filings specific to any Global Development Plan activity (for clarity, excluding any NDA, MAA or Regulatory Approval); and (ii) notwithstanding the foregoing limitation of Global Development Costs to external costs and expenses, the Cost of Goods of Licensed Product used in the conduct of such Global Development Plan activity. Notwithstanding the foregoing or any other provision of this Agreement to the contrary, Global Development Costs shall exclude the Cost of Goods of any validation batches of Licensed Product that […***…] (any such additional validation batch, an “Excluded Validation Batch”).
1.60 “Global Development Plan” shall mean the written plan attached hereto as Exhibit B for the conduct of (a) the Lead Indication Trials; (b) the GLP Studies specified therein; and (c) the CMC development activities specified therein; in each case of (a) to (c), to be conducted by or on behalf of Cidara (but which may include activities in the Mundipharma Territory that will be conducted by Mundipharma and/or its Affiliates) that are intended to support the clinical development of, and filing of XXXx for, the Lead Indication Product in the Lead Indications in the Major Markets, as such plan may be amended from time to time in accordance with Section 3.1 and Section 4.1(b); provided, however, that the Global Development Plan shall not include the conduct of (i) the Lead Indication Trial in the Prophylaxis Indication in China, and (ii) the European Pediatric Investigational Plan referred to in the Mundipharma Territory Development Plan.
1.61 “GLP” shall mean current good laboratory practices as established by the FDA and as interpreted by relevant ICH guidelines; in each case, as amended from time to time.
1.62 “GLP Study” shall mean any non‑clinical study of a Compound or Product (a) that is intended to comply with GLP or (b) the results of which would be required to be reported to any Regulatory Authority.
1.63 “GMP” shall mean current good manufacturing practices and standards for the production of drugs and finished pharmaceuticals, as set forth in 21 C.F.R. Parts 210 and 211, as amended from time to time and as interpreted by relevant ICH guidelines.
1.64 “GVP” shall mean the current good pharmacovigilance practices guidelines that are mandatory to follow for pharmaceutical products marketed in the EU.
1.65 “Grant-Back License” shall mean the licenses granted by Mundipharma to Cidara pursuant to Section 2.6.
1.66 “ICH” shall mean the International Council for Harmonisation (formerly the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use).
8.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
1.67 “IND” shall mean an investigational new drug application, clinical trial application, clinical trial exemption, or similar application or submission filed with or submitted to a Regulatory Authority in a jurisdiction that is necessary to commence human clinical trials in such jurisdiction, including any such application filed with the FDA pursuant to 21 C.F.R. Part 312.
1.68 “Independent Development Party” shall have the meaning provided in Section 4.3(b).
1.69 “Information” shall mean any and all tangible and intangible (a) techniques, technology, practices, trade secrets, inventions (whether patentable or not), methods, knowledge, know-how, data (including biological, chemical, pharmacological, toxicological, safety, pharmacokinetic, clinical, CMC, analytical, quality control, mechanical, software, electronic and other data), results of research, preclinical and non-clinical studies (including in vitro, in vivo, and ex vivo studies), clinical trials and other testing, software and algorithms, and (b) compositions of matter, cells, cell lines, assays, animal models and physical, biological or chemical material; that, in each case, are not in the public domain.
1.70 “Insolvency Event” shall mean, with respect to a party, circumstances under which such party: (a) makes a general assignment for the benefit of, or an arrangement or composition generally with, its creditors; (b) appoints or suffers appointment of an examiner or of a receiver, custodian, liquidator, trustee or similar person over all or substantially all of its property; (c) passes a resolution for its winding up, liquidation, dissolution, reorganization or similar process (but excluding (i) any such process for the purpose of, or in connection with, any solvent amalgamation, solvent reconstruction or solvent restructuring, (ii) a consolidation with a wholly-owned subsidiary of such party, (iii) a merger effected exclusively to change the domicile of such party and (iv) any transaction or series of transactions principally for bona fide equity financing purposes in which cash is received by such party or indebtedness of such party is cancelled or converted or a combination thereof); or (d) files a petition or commences a proceeding under any bankruptcy or insolvency act or law or has any such involuntary petition filed, or involuntary proceeding commenced, against it, unless, in the case of any such involuntary petition or involuntary proceeding, such petition or proceeding is dismissed within 90 days after the filing or commencement thereof.
1.71 “Invention” shall mean any invention or discovery, whether or not patentable, that is made, conceived, generated or reduced to practice, in whole or in part, in the course and as a result of the conduct of the activities expressly contemplated by this Agreement.
1.72 “Joint Commercialization Committee” or “JCC” shall have the meaning provided in Section 3.2.
1.73 “Joint Development Committee” or “JDC” shall have the meaning provided in Section 3.2.
1.74 “Joint Invention” shall mean any Invention made jointly by, on the one hand, one or more employees, consultants or contractors of Mundipharma and/or any of its Affiliates, and, on the other hand, one or more employees, consultants or contractors of Cidara.
9.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
1.75 “Joint Know-How” shall mean any Information and Data made or generated jointly by, on the one hand, one or more employees, consultants or contractors of Mundipharma and/or any of its Affiliates, and, on the other hand, one or more employees, consultants or contractors of Cidara and/or any Cidara-Controlled Affiliates.
1.76 “Joint Patents” shall mean Patents claiming Joint Inventions.
1.77 “Joint Steering Committee” or “JSC” shall have the meaning provided in Section 3.1(a).
1.78 “Joint Technology” shall mean the Joint Patents, Joint Inventions and Joint Know-How.
1.79 “JSC Dispute Resolution Matrix” shall mean Exhibit C hereto.
1.80 “Key Matter” shall have the meaning provided in Section 3.1(f).
1.81 “Knowledge” means (i) the actual knowledge of the executive officers and outside intellectual property counsel of Cidara; and (ii) the knowledge that any of such individuals reasonably should have gained through operating in the ordinary course of business.
1.82 […***…].
1.83 “Lead Compound” shall mean the active pharmaceutical ingredient of rezafungin with chemical formula: N5.1,6-anhydro[(4R,5R)-4-hydroxy-2-[34- (pentyloxy)[11,21:24,31-terphenyl]-14-carboxamido]-5-[2- (trimethylazaniumyl)ethyl]-L-ornithyl-L-threonyl-trans-4-hydroxy-L-prolyl-(4S)-4-hydroxy-4-(4-hydroxyphenyl)-L-threonyl-L-threonyl-(3S,4S)-3-hydroxy-4-methyl-L-proline] acetate, having the chemical structure set forth in Exhibit E attached hereto.
1.84 “Lead Indication Product” shall mean the Licensed Product containing the Lead Compound in a formulation for intravenous administration that is being investigated in the Lead Indication Trials for the Lead Indications as of the Effective Date.
1.85 “Lead Indications” shall mean: (a) the treatment of candidemia and/or invasive candidiasis (the “Treatment Indication”); and (b) the prophylaxis of invasive fungal infections in adult allogeneic blood and marrow transplant recipients (the “Prophylaxis Indication”).
1.86 “Lead Indication Trials” shall mean the ReSTORE Trial and the ReSPECT Trial.
1.87 “License” shall have the meaning provided in Section 2.1(b).
1.88 “Licensed Product” shall mean any Product in a formulation for intravenous administration, including all uses, presentations and dosage strengths thereof. For clarity, Licensed Product shall, subject to Section 4.3, include an Expanded Licensed Product.
10.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
1.89 “Licensed Product Global Development Expansion” shall have the meaning provided in Section 4.3(a).
1.90 “MAA” shall mean an application or submission for approval to market a pharmaceutical product filed with the governing Regulatory Authority in any jurisdiction other than the U.S., including, without limitation, a marketing authorisation application filed with the EMA using the centralized EU filing procedure or filed with the applicable national Regulatory Authority in an individual European country, whether or not such country is an EU member state (including, for purposes of this Agreement, the United Kingdom) if the centralized EU filing procedure is not used, or is not available for use, in such European country.
1.91 “Major European Country” shall mean any of the following countries: France, Germany, Italy, Spain and the United Kingdom (without regard to whether or not any of the foregoing is an EU member state at any given time).
1.92 “Major Market” shall mean a […***…].
1.93 “Manufacturing License” shall have the meaning provided in Section 2.1(b).
1.94 “Marketing Partner” shall mean a Third Party to which Mundipharma or its Affiliates has granted the right solely to market or promote, but not to sell or offer for sale, Licensed Product in the Field in the Mundipharma Territory.
1.95 “Material Impact” shall mean (a) a material adverse impact on (i) the likelihood or timing of obtaining Regulatory Approval (excluding pricing and reimbursement approval) of Licensed Product in a party’s Territory, (ii) continuing maintenance of the Regulatory Approval (excluding pricing and reimbursement approval) of Licensed Product in a party’s Territory and/or (iii) the safety profile of Licensed Product, or (b) an unacceptable risk to patient/subject safety.
1.96 “Material Impact Objection” shall have the meaning provided in Section 3.1(f)(iii).
1.97 “MA Variation” shall mean an application for a variation or amendment to an MAA filed with the governing Regulatory Authority in any jurisdiction other than the U.S., including, without limitation, an application for variation to a marketing authorisation application filed with the EMA using the centralized EU filing procedure or filed with the applicable national Regulatory Authority in an individual European country, whether or not such country is an EU member state (including, for purposes of this Agreement, the United Kingdom) if the centralized EU filing procedure is not used, or is not available for use, in such European country.
1.98 “Mundipharma” shall have the meaning first set out above.
1.99 “Mundipharma Invention” shall mean any Invention made solely by one or more employees, consultants or contractors of Mundipharma or any of its Affiliates.
1.100 “Mundipharma Know-How” shall mean all Information and Data that: (a) is generated, developed or obtained by or on behalf of Mundipharma or any of its Affiliates or
11.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
Sublicensees during the Term in the development, registration, manufacture, use or commercialization of Compound or Licensed Product; or (b) is otherwise Controlled by Mundipharma or any of its Affiliates during the Term and is necessary for, or is both useful for and actually used by Mundipharma or any of its Affiliates or Sublicensees in, the development, registration, manufacture, use or commercialization of Compound or Licensed Product; in each case, including Mundipharma Inventions; but, in each case, excluding: (i) Expanded Licensed Product Clinical Efficacy Data generated by or on behalf of Mundipharma as an Independent Development Party unless the parties have executed an Expanded Licensed Product Amendment with respect to the applicable Expanded Licensed Product; (ii) Mundipharma Patents; and (iii) Joint Technology.
1.101 “Mundipharma Marketing Information” shall mean that portion of the Mundipharma Know-How constituting strategic marketing information, strategic pricing information, marketing materials, marketing know-how, data and information resulting from health economic outcomes research, real world evidence and observational studies, market research and marketing advisory boards, and any other studies, research or Third Party consultancy performed to support pricing or reimbursement approval but not necessary to obtain or maintain Regulatory Approvals other than pricing and reimbursement approvals.
1.102 “Mundipharma Patents” shall mean: (a) all Patents claiming Mundipharma Inventions; and (b) all other Patents Controlled by Mundipharma or any of its Affiliates that claim inventions that are necessary for, or both useful for and actually used by or on behalf of Mundipharma or any of its Affiliates or Sublicensees in, the development, registration, manufacture, use or commercialization of Compound or Licensed Product; but, in each case, excluding Joint Patents.
1.103 “Mundipharma Peak Incremental Net Sales Forecast” shall have the meaning provided in Section 4.3(a)(i).
1.104 “Mundipharma Subcutaneous Peak Net Sales Forecast” shall have the meaning provided in Section 4.4(c)(ii).
1.105 “Mundipharma Technology” shall mean the Mundipharma Patents, Mundipharma Inventions and Mundipharma Know-How.
1.106 “Mundipharma Territory” shall mean the entire world, excluding the Cidara Territory.
1.107 “Mundipharma Territory Plan” shall mean the written plan attached hereto as Exhibit F setting forth the Mundipharma Territory-Specific clinical trials and GLP Studies of the Lead Indication Product in the Lead Indications to be conducted by or on behalf of Mundipharma in the Mundipharma Territory that are required to support MAA filing and Regulatory Approval (excluding pricing and reimbursement approval) for the Lead Indication Product in the Lead Indications in the Mundipharma Territory, including the European Paediatric Investigational Plan, as such plan may be amended from time to time in accordance with Section 3.1 and Section 4.2.
12.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
1.108 “Mundipharma Territory-Specific” shall mean, in reference to any GLP Study or clinical trial of Lead Indication Product in a Lead Indication, or of Pediatric Licensed Product, in each case, in the Mundipharma Territory, that such study or trial is necessary to support MAA filing and Regulatory Approval of Lead Indication Product in such Lead Indication or Pediatric Licensed Product in the Mundipharma Territory or any portion thereof, but is not included in the Global Development Plan.
1.109 “NDA” shall mean a New Drug Application (as more fully defined in 21 CFR 314.5, et seq.) filed with the FDA, or any successor application thereto in the U.S.
1.110 “Net Sales” shall mean the gross amounts amount billed or invoiced by Mundipharma, its Affiliates and Sublicensees (in each case, a “Selling Party”) for sales or other dispositions of Licensed Products to Third Parties (excluding Sublicensees), less the following amounts specifically attributable to Licensed Products and actually incurred, allowed, paid or accrued, or otherwise specifically allocated to Licensed Products by the Selling Party (if not previously deducted in calculating the amount invoiced), all in compliance with applicable Accounting Standards, consistently applied by the Selling Party:
(a) Normal and customary trade discounts, including trade, cash and quantity discounts or trade rebates, credits or refunds, actually allowed or taken (including amounts repaid, discounted or credited by reason of risk sharing schemes with any governmental authority or Regulatory Authority); credits or allowances actually granted or made for rejection of or return of previously sold Licensed Products, including recalls, or for retroactive price reductions and billing errors;
(b) […***…];
(c) governmental and other rebates (or chargeback payments, credits or other equivalents thereof, including amounts repaid, discounted or credited by reason of retroactive price reductions, discounts, or rebates, which are, in each case, imposed upon Mundipharma, its Affiliates or Sublicensees by any governmental authority or Regulatory Authority) actually granted to managed health care organizations, commercial insurance companies, pharmacy benefit managers (or equivalents thereof), distributors, governments, their agencies and purchasers, and reimbursers;
(d) charges separately invoiced for outbound freight, insurance, transportation, postage and handling; and
(e) tariffs, customs duties and Sales Tax levied on the billing amount for Licensed Products or otherwise imposed with respect to the sale, transportation or delivery of Licensed Products, and, in each case, actually paid, as adjusted for rebates and refunds;
provided that, in each case ((a) through (f)), (1) each such deduction is calculated in a manner consistent with the Selling Party’s customary practice for pharmaceutical products and in accordance with applicable Accounting Standards, consistently applied by the Selling Party, (2) each such deduction is directly allocable to Licensed Product, or apportioned on a good faith, fair and equitable basis to Licensed Product and other products of the Selling Party and its Affiliates such that Licensed
13.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
Product does not bear a disproportionate portion of such deductions, and (3) no particular amount identified above shall be deducted more than once in calculating Net Sales (i.e., no “double counting” of deductions).
For clarification, sale or other disposition of Licensed Product by a Selling Party to another Selling Party for resale by such other Selling Party to a Third Party (other than a Selling Party) shall not be deemed a sale for purposes of this definition of “Net Sales,” provided that the subsequent resale is included in the computation of Net Sales. In the event of any sale or other disposition of Licensed Product for any consideration other than exclusively monetary consideration on bona fide arm’s-length terms (including any sale or other disposition of Licensed Product by a Selling Party to another Selling Party for end use by such other Selling Party), then for purposes of calculating Net Sales under this Agreement, such Licensed Product shall be deemed to have been sold exclusively for cash at the weighted (by sales volume) average sale price of such Licensed Product in bona fide arm’s-length transactions (when sold alone, and not with other products) in the applicable country in which such sale or other disposition occurred during the applicable accounting period. Transfers or dispositions of Licensed Product for charitable, research and development, clinical or humanitarian purposes, in all cases without consideration, shall be disregarded in determining Net Sales.
On a country-by-country basis, if a Licensed Product is sold in a country as part of a Combination Product in a calendar quarter, Net Sales of such Licensed Product in such country during such calendar quarter for the purpose of determining royalties and commercialization milestone payments due hereunder shall be calculated as follows:
(i) In the event that both (x) a Single-Agent Licensed Product is sold separately in finished form in such country during such calendar quarter and (y) the Other Active(s) in such Combination Product are sold separately in finished form in such country during such calendar quarter, then Net Sales of such Licensed Product shall be determined by multiplying the actual Net Sales of the Combination Product calculated pursuant to the preceding provisions of this Section 1.110 (“Actual Combination Product Net Sales”) in such country during such calendar quarter by the fraction, A / (A+B) where A is the weighted average sale price of such Single-Agent Licensed Product when sold separately in finished form in such country during such calendar quarter, and B is the weighted average sale price of the Other Active(s) in the Combination Product when sold separately in finished form in such country during such calendar quarter.
(ii) In the event that a Single-Agent Licensed Product is sold separately in finished form in such country during such calendar quarter, but the Other Active(s) in such Combination Product are not sold separately in finished form in such country during such calendar quarter, then Net Sales of such Licensed Product shall be calculated by multiplying the Actual Combination Product Net Sales of the Combination Product in such country during such calendar quarter by the fraction A / C where A is the weighted average sale price of such Single-Agent Licensed Product when sold separately in finished form in such country during such calendar quarter and C is the weighted average sale price of the Combination Product in such country during such calendar quarter.
14.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
(iii) In the event that no Single-Agent Licensed Product is sold separately in finished form in such country during such calendar quarter, but the Other Active(s) in such Combination Product are sold separately in finished form in such country during such calendar quarter, Net Sales of such Licensed Product shall be calculated by multiplying the Actual Combination Product Net Sales of the Combination Product by the fraction (C‑B) / C, where B is the weighted average sale price of the Other Active(s) in the Combination Product when sold separately in finished form in such country during such calendar quarter, and C is the weighted average sale price of the Combination Product in such country during such calendar quarter.
(iv) In the event that neither any Single-Agent Licensed Product is sold separately in finished form in such country during such calendar quarter, nor the Other Active(s) in such Combination Product are sold separately in finished form in such country during such calendar quarter, then the methodology for determining Net Sales of such Licensed Product in such country shall be mutually agreed in writing by the parties in good faith based on the relative contributions of the Compound and the Other Active(s) in such Combination Product to the total value of the Combination Product.
1.111 “New Indication” shall have the meaning provided in Section 4.3(a).
1.112 “New Other Product Indication” shall have the meaning provided in Section 4.5(c).
1.113 “NIH Trial” shall have the meaning provided in Section 4.4(a).
1.114 “NMPA” shall mean China’s National Medical Products Administration or any successor agency thereto in China having substantially the same function.
1.115 “Non-Exclusively Licensed Mundipharma Patent Claim” shall mean a claim of a Mundipharma Patent that covers:
(a) […***…]; or
(b) […***…].
1.116 “Other Active” shall mean any active pharmaceutical ingredient other than Compound.
1.117 “Other Product” shall have the meaning provided in Section 4.5(a).
1.118 “Other Product Amendment” shall have the meaning provided in Section 4.5(c).
1.119 “Other Product Efficacy Trial” shall have the meaning provided in Section 4.5(b).
1.120 “Other Product Efficacy Trial Notice” shall have the meaning provided in Section 4.5(b).
15.
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
1.121 “Other Product Exercise Notice” shall have the meaning provided in Section 4.5(c).
1.122 “Other Product Option” shall have the meaning provided in Section 4.5.
1.123 “Other Product Option Period” shall mean the period beginning on the Effective Date and, subject to earlier termination of this Agreement, expiring […***…] after […***…].
1.124 “Patents” shall mean (a) all national, regional and international patents and patent applications filed in any country or jurisdiction, including without limitation provisional patent applications, (b) all patent applications filed either from such patents and patent applications or from a patent application claiming priority from either of these, including any continuation,
continuation-in-part, division, provisional, converted provisional and continued prosecution applications, or any substitute applications, (c) any patent issued with respect to or in the future issued from any such patent applications including utility models, xxxxx patents and design patents and certificates of invention, and (d) any and all extensions or restorations by existing or future extension or restoration mechanisms, including revalidations, reissues, reexaminations and extensions (including any supplementary protection certificates, patent term extensions and the like) of the foregoing patents or patent applications.
1.125 “Peak Incremental Net Sales Forecasts” shall have the meaning provided in Section 4.3(a)(i).
1.126 “Pediatric Licensed Product” shall mean Licensed Product intended for use in patients younger than 18 years of age.
1.127 “Person” any natural person or Entity.
1.128 “Phase 2 Trial” shall mean a human clinical trial that would satisfy the requirements for a Phase 2 study as defined in 21 CFR § 312.21(b) (or any amended or successor regulations), regardless of where such clinical trial is conducted.
1.129 “Phase 3 Trial” shall mean a human clinical trial that would satisfy the requirements for a Phase 3 study as defined in 21 CFR § 312.21(c) (or any amended or successor regulations), regardless of where such clinical trial is conducted.
1.130 “Prior CDA” shall mean that certain Mutual Non-Disclosure Agreement between Cidara and Mundipharma International Limited dated December 14, 2018.
1.131 “Product” shall mean any pharmaceutical composition or preparation containing or comprising Compound (whether or not as the sole active ingredient), in any formulation, including all uses, routes of administration, presentations and dosage strengths thereof.
1.132 “Product Filings” shall mean all INDs, NDAs, XXXx, MA Variations, Regulatory Approvals, and other filings with, and formal submissions to, Regulatory Authorities, in each case, with respect to the development, manufacture, and marketing and sale of Licensed Product in any country or other jurisdiction.
1.133 “Product Trademarks” shall have the meaning provided in Section 8.8(a).
1.134 “Prophylaxis Indication” shall have the meaning provided in Section 1.85.
1.135 “Receiving Party” shall have the meaning provided in Section 7.1.
1.136 “ReCoRD” shall mean a document prepared by Mundipharma in respect of each MAA approved in each country in the Mundipharma Territory, together with XXXx approved by the European Commission and NMPA, with a unique identification number, which comprises relevant registered compliance data from Module 3 of the relevant MAA.
1.137 “Regulatory Approval” shall mean, with respect to a pharmaceutical product in a particular jurisdiction, all approvals or other permissions from the applicable Regulatory Authority in such jurisdiction necessary to develop, market and sell such product in such jurisdiction, including approvals of INDs, NDAs, XXXx, MA Variations, and pricing and reimbursement approvals if required for marketing or sale of such product in such jurisdiction.
1.138 “Regulatory Authority” shall mean any country, federal, supranational, state or local regulatory agency, department, bureau or other governmental or regulatory authority having the administrative authority to regulate the development, marketing or sale of pharmaceutical products in any country or other jurisdiction, including the pricing and reimbursement of such products, and other market access activities.
1.139 “Regulatory Exclusivity” shall mean any exclusive marketing rights or data exclusivity rights conferred by any Regulatory Authority with respect to a pharmaceutical product other than a Patent, including orphan drug exclusivity, new chemical entity exclusivity, data exclusivity, or pediatric exclusivity.
1.140 “ReSPECT Trial” shall mean the Phase 3 Trial of the Lead Indication Product in the Prophylaxis Indication described in Cidara Clinical Protocol No. CD101.IV.3.08, entitled “A Phase 3, Multicenter, Randomized, Double-blind Study of the Efficacy and Safety of Rezafungin for Injection Versus the Standard Antimicrobial Regimen to Prevent Invasive Fungal Diseases in Adults Undergoing Allogeneic Blood and Marrow Transplantation,” as amended from time to time in accordance with this Agreement.
1.141 “ReSTORE Trial” shall mean the Phase 3 Trial of the Lead Indication Product in the Treatment Indication described in Cidara Clinical Protocol No. CD101.IV.3.05, entitled “A Phase 3, Multicenter, Randomized, Double-blind Study of the Efficacy and Safety of Rezafungin for Injection Versus Intravenous Caspofungin Followed by Optional Oral Fluconazole Step-down in the Treatment of Subjects with Candidemia and/or Invasive Candidiasis,” as amended from time to time in accordance with this Agreement.
1.142 “Right of Reference” shall mean: (a) in the U.S., a “right of reference or use,” as such term is defined in 21 C.F.R. 314.3(b); or (b) in any other country or jurisdiction, the equivalent authority to rely upon, and otherwise use, an investigation for the purpose of filing, and conducting a clinical trial under, an IND, or obtaining approval of an NDA, MAA, MA Variation or other Regulatory Approval, including the ability to make available the underlying raw data from the investigation for audit by the applicable Regulatory Authority in such country or other jurisdiction, if necessary.
1.143 “Royalty Term” shall have the meaning provided in Section 5.8.
1.144 “Sales Tax” shall have the meaning provided in Section 6.3(d).
1.145 “Selling Party” shall have the meaning set forth in Section 1.110.
1.146 “Single-Agent Product” shall mean a Licensed Product containing Compound as its sole active pharmaceutical ingredient.
1.147 “Subcommittee” shall mean the JDC, the JMC, the JCC or any other subcommittee established by the JSC pursuant to Section 3.2.
1.148 “Subcutaneous Efficacy Trial” shall have the meaning provided in Section 4.4(b).
1.149 “Subcutaneous Efficacy Trial Notice” shall have the meaning provided in Section 4.4(b).
1.150 “Subcutaneous Global Development Plan” shall mean, solely in the event of Mundipharma’s exercise of the Subcutaneous Product Option, a written plan for the conduct of Cidara-sponsored clinical trial(s) and GLP Studies of Subcutaneous Product in the Subcutaneous Indication(s), and CMC development activities to be conducted by or on behalf of Cidara (which may include activities in the Mundipharma Territory that will be performed by Mundipharma and/or its Affiliates) in support of the clinical development of, and filing of XXXx for, the Subcutaneous Product in the Subcutaneous Indication(s) in the Major Markets.
1.151 “Subcutaneous Indication” shall mean an indication in the Field that is the subject of a Subcutaneous Product Amendment executed by the parties.
1.152 “Subcutaneous License Expansion” shall have the meaning provided in Section 4.4(c)(i).
1.153 “Subcutaneous Mundipharma Territory Plan” shall mean, solely in the event of Mundipharma’s exercise of the Subcutaneous Product Option, a written plan setting forth the Mundipharma Territory-Specific (mutatis mutandis) clinical trials and GLP Studies of Subcutaneous Product in the Subcutaneous Indication to be conducted by or on behalf of Mundipharma in the Mundipharma Territory that are required to support MAA filing and Regulatory Approval (excluding pricing and reimbursement approval) for Subcutaneous Product in the Subcutaneous Indication in the Mundipharma Territory but are not included in the Subcutaneous Global Development Plan.
1.154 “Subcutaneous Negotiation Period” shall have the meaning provided in Section 4.4(c).
1.155 “Subcutaneous Option Payment” shall have the meaning provided in Section 4.4(c)(ii).
1.156 “Subcutaneous Peak Incremental Net Sales Forecasts” shall have the meaning provided in Section 4.4(c)(ii).
1.157 “Subcutaneous Product” shall mean any Product in a formulation for subcutaneous administration, including all uses, presentations and dosage strengths thereof.
1.158 “Subcutaneous Product Amendment” shall have the meaning provided in Section 4.4(c).
1.159 “Subcutaneous Product Option” shall have the meaning provided in Section 4.4(a).
1.160 “Subcutaneous Product Option Period” shall mean the period beginning on the Effective Date and, subject to earlier termination of this Agreement, expiring […***…] after the latest to occur of […***…].
1.161 “Sublicense” shall mean (a) a sublicense under the License or any portion thereof, or (b) a right to market, promote and sell Licensed Product in the Field in the Mundipharma Territory. As used in this Agreement, a Sublicense shall not include the rights granted to a Distributor or to a Marketing Partner.
1.162 “Sublicensee” shall mean any Affiliate or Third Party that has received a Sublicense, directly or indirectly through one or more tiers, from Mundipharma or its Affiliate. As used in this Agreement, “Sublicensee” shall not include a Distributor or a Marketing Partner.
1.163 “Successful Completion” shall mean:
(a) with respect to the ReSTORE Trial, delivery by Cidara to Mundipharma of top-line results of the ReSTORE Trial with results in the assessment of Global Cure at […***…] of subjects who are randomized to Licensed Product compared to subjects who are randomized to intravenous caspofungin; and
(b) with respect to the ReSPECT Trial, delivery by Cidara to Mundipharma of top-line results of the ReSPECT Trial with results in the assessment of Fungal-Free Survival at […***…] of subjects who are randomized to Licensed Product compared to subjects randomized to the standard antimicrobial regimen.
1.164 “Supply Price” shall have the meaning provided in Section 4.18(d).
1.165 “Taxes” shall have the meaning provided in Section 6.3(b).
1.166 “Territory” shall mean, as applicable, the Cidara Territory or the Mundipharma Territory.
1.167 “Term” shall have the meaning provided in Section 10.1.
1.168 “Third Party” shall mean any entity other than Cidara or Mundipharma or an Affiliate of Cidara or Mundipharma.
1.169 “Treatment Indication” shall have the meaning provided in Section 1.85.
1.170 “U.S.” shall mean the United States of America, including its territories and possessions.
1.171 “Valid Claim” shall mean: (a) a claim of an issued and unexpired patent, or a supplementary protection certificate thereof, which has not been held permanently revoked, unenforceable or invalid by a decision of a court, patent office or other forum of competent jurisdiction, unappealable or unappealed within the time allowed for appeal and that is not admitted to be invalid or unenforceable through reissue, disclaimer or otherwise; or (b) a claim of a pending patent application that has not been abandoned, finally rejected or expired without the possibility of appeal or re-filing and that has not been pending for more than […***…] years from the filing date of the earliest patent application from which such claim derives priority; […***…].
2. | LICENSE GRANTS |
2.1 License Grant to Mundipharma. Subject to the terms and conditions of this Agreement, Cidara hereby grants to Mundipharma, during the Term:
(a) an exclusive (even as to Cidara and its Affiliates, except as set forth in Section 2.4), royalty‑bearing license, including the right to sublicense as expressly permitted by Section 2.2, under the Cidara Technology and Cidara’s interest in Joint Technology, solely to develop, register, use, sell, have sold, offer for sale and import Licensed Product in the Field in the Mundipharma Territory (the “Exclusive License”); provided, however, that for purposes of the Exclusive License, the Cidara Technology excludes all Cidara Patents existing outside of the Mundipharma Territory; and
(b) a co‑exclusive (with Cidara), royalty-bearing license (but solely during the Royalty Term and pursuant to and in accordance with Section 5.3 and Sections 5.7 through 0), without the right to sublicense (other than to its Affiliates pursuant to Section 2.2), but, for clarity, with the right to subcontract to a CMO, under the Cidara Technology and Cidara’s interest in Joint Technology, to make and have made Compound solely for incorporation into Licensed Products, and to make and have made Licensed Products; in each case, anywhere in the world but solely to develop, register, use, sell, have sold, offer for sale and import such Licensed Products in the Field in the Mundipharma Territory (the “Manufacturing License” and, collectively with the Exclusive License, the “License”).
2.2 Sublicenses and Appointments. The License shall include:
(a) the right to grant Sublicenses (through one or more tiers) to any of the Affiliates of Mundipharma without Cidara’s consent, provided that any further Sublicense proposed to be granted by any such Affiliate to a Third Party in a Major Market shall be subject to Section 2.2(b);
(b) the right to grant Sublicenses to a Third Party in a Major Market only with Cidara’s prior written consent, not to be unreasonably withheld, conditioned or delayed;
(c) the right to grant Sublicenses to a Third Party outside the Major Markets in the Mundipharma Territory without Cidara’s prior written consent;
(d) the right to appoint Distributors and Marketing Partners, in each case, without Cidara’s prior written consent; and
(e) the right to appoint any other Third Party subcontractors (and grant limited sublicenses as necessary in connection with such subcontracting) to perform Mundipharma’s obligations and/or to exercise Mundipharma’s rights herein on behalf of Mundipharma and/or its Affiliates without Cidara’s consent.
Any Sublicense granted to any Affiliate of Mundipharma or to any Third Party, and any appointment of a Marketing Partner or Distributor shall be in writing and shall be subject to, and consistent with, the terms and conditions of this Agreement. Mundipharma shall be fully responsible for the compliance of its Affiliates, Sublicensees, Marketing Partners, Distributors, other subcontractors (including any CMO contracted by Mundipharma or its Affiliate to package and label Licensed Product manufactured and supplied by or on behalf of Cidara), with the terms and conditions of this Agreement and shall remain solely liable for the performance of its obligations hereunder, notwithstanding any such Sublicense or appointment. Mundipharma shall promptly notify Cidara in writing of the execution of any Sublicense agreement with a Third Party and shall provide Cidara with a copy of such Sublicense agreement, and any amendment thereto, no later than 30 days following execution thereof; provided, that Mundipharma may redact any confidential or financial information contained therein that is unnecessary for Cidara to ascertain compliance with this Agreement.
2.3 Initial Delivery of Cidara Know-How; Ongoing Know-How Exchange.
(a) Within 30 days after the Effective Date, Cidara shall, at no additional charge to Mundipharma, deliver to Mundipharma or its designated Affiliate such existing and available (in recorded form) Cidara Know-How in the possession of Cidara and/or Cidara-Controlled Affiliates as is necessary or useful for Mundipharma and its Affiliates to (i) exercise the License in accordance with this Agreement; and (ii) otherwise exercise Mundipharma’s rights and perform Mundipharma’s obligations under this Agreement. Thereafter, on an ongoing basis during the Term, Cidara shall also disclose to Mundipharma or its designated Affiliate such additional Cidara Know-How arising after the Effective Date as is necessary or useful for Mundipharma and its Affiliates to (i) exercise the License in accordance with this Agreement and (ii) otherwise exercise Mundipharma’s rights and perform Mundipharma’s obligations under this Agreement. Without limiting the generality of the foregoing, Cidara shall provide to Mundipharma true and complete copies of all written, graphic or electronic embodiments of Data generated by or on behalf of Cidara or any of its Affiliates or licensees, including, without limitation, all draft and final protocols and all final reports of any GLP Study or clinical trial of Compound or Product in the Field conducted by or on behalf of Cidara and/or Cidara-Controlled Affiliates, and all pharmacology, toxicology, pharmacokinetic and other data with respect to Compound or Licensed Product, and Mundipharma shall have the right to use such disclosed Data solely within the scope of the License and as otherwise expressly permitted by this Agreement. Notwithstanding the foregoing, Mundipharma shall have no license or right to use any Expanded Licensed Product Clinical Efficacy Data generated by or on behalf of Cidara and/or Cidara-Controlled Affiliates in support of any MAA, MA Variation or other Product Filing or Regulatory Approval for the applicable Expanded Licensed Product in the Mundipharma Territory or in the commercialization of an Expanded Licensed Product in the Mundipharma Territory, unless the parties have executed an Expanded Licensed Product Amendment for such Expanded Licensed Product and, if applicable, Mundipharma has paid the applicable Expanded Licensed Product Buy‑In Fee with respect thereto.
(b) On an ongoing basis during the Term, Mundipharma shall disclose to Cidara such Mundipharma Know-How as is necessary or useful for Cidara to (i) exercise the Grant-Back License in accordance with this Agreement or (ii) otherwise exercise Cidara’s rights and perform Cidara’s obligations under Article 4 of this Agreement. Without limiting the generality of the foregoing, Mundipharma shall provide to Cidara true and complete copies of all written, graphic or electronic embodiments of Data generated by or on behalf of Mundipharma or any of its Affiliates or Sublicensees, including, without limitation, all draft and final protocols and final reports of any GLP Study or clinical trial of Compound or Licensed Product (including the trial described in the European Paediatric Investigational Plan) in the Field conducted by or on behalf of Mundipharma and/or its Affiliates, and all pharmacology, toxicology, pharmacokinetic and other data with respect to Compound or Licensed Product, and Cidara shall have the right to use such disclosed Data solely within the scope of the Grant-Back License and as otherwise expressly permitted by this Agreement. Notwithstanding the foregoing, Cidara shall have no license or right to use any Expanded Licensed Product Clinical Efficacy Data generated by or on behalf of Mundipharma and/or its Affiliates in support of any NDA, MAA, MA Variation or other Product Filing or Regulatory Approval for the applicable Expanded Licensed Product in the Cidara Territory or in the commercialization of such Expanded Licensed Product in the Cidara Territory, unless the parties have executed an Expanded Licensed Product Amendment for such Expanded Licensed Product and Cidara has paid the applicable Expanded Licensed Product Buy‑In Fee with respect thereto.
2.4 Retained Rights.
(a) Under Exclusive License. Notwithstanding the exclusivity of the Exclusive License, Cidara retains the non‑exclusive right to practice the Cidara Technology and Cidara’s interest in Joint Technology in the Mundipharma Territory for the purpose of performing development activities related to the Licensed Product, including Global Development Plan activities, in the Mundipharma Territory, including the right to file and maintain INDs for, and to use, Licensed Product for such purposes in the Mundipharma Territory.
(b) Under Manufacturing License. Notwithstanding the co‑exclusivity of the Manufacturing License, Cidara retains the right to grant licenses under the Cidara Technology and Cidara’s interest in Joint Technology to its licensees that have the right to market, promote and sell Licensed Product in the Cidara Territory, to make and have made Compound for incorporation in Licensed Product, and to make and have made Licensed Product, anywhere in the world, subject to the terms and conditions of this Agreement.
(c) Development. For clarity, Cidara shall at all times have the right to develop Compound and Product in the Field anywhere in the world, subject to compliance with its obligations under Articles 3 and 4 hereof.
2.5 Mundipharma Negative Covenants. Mundipharma hereby covenants on behalf of itself and its Affiliates not to practice, and not to permit or cause any Affiliate, Sublicensee, Marketing Partner, Distributor or other Third Party to practice, any Cidara Technology for any purpose other than as expressly authorized in this Agreement. Mundipharma further covenants on behalf of itself and its Affiliates:
(a) not to develop, register, use, sell, have sold or offer for sale or seek Regulatory Approval for Compound or Product (including Licensed Product) in the Cidara Territory. Without limiting the generality of the foregoing, Mundipharma hereby covenants on behalf of itself and its Affiliates not to, and not to permit or cause any Affiliate to, sell or provide Compound or Licensed Product to any Sublicensee, Distributor or other Third Party if any of them knows that Compound or Licensed Product sold or provided to such Third Party would be sold or transferred, directly or indirectly, for use in the Cidara Territory;
(b) except as otherwise mutually agreed by the parties in writing pursuant to Section 4.3, Section 4.4 or Section 4.5, not to develop, register, use, sell, have sold, offer for sale or import or seek Regulatory Approval for (i) any Licensed Product containing any Compound other than the Lead Compound, (ii) any Licensed Product containing the Lead Compound other than the Lead Indication Product and the Pediatric Licensed Product for the Mundipharma Territory; (iii) any Combination Product, or (iv) any Product other than Licensed Product;
(c) not to conduct or have conducted any GLP Study or clinical trial of Compound or Licensed Product, except in accordance with a protocol approved in accordance with Article 3 and the JSC Dispute Resolution Matrix;
(d) not to make or have made Compound or Product, except Compound and Licensed Product within the express scope of the Manufacturing License; and
(e) not to grant, or purport to grant, any Affiliate of Mundipharma or any Third Party any license or other right to do any of the foregoing.
2.6 License Grant-Back to Cidara.
(a) Cidara Territory. Subject to the terms and conditions of this Agreement, Mundipharma hereby grants to Cidara:
(i) an exclusive (even as to Mundipharma), royalty‑free, fully‑paid, irrevocable, perpetual license, with the right to sublicense through multiple tiers of sublicense, under the Mundipharma Technology (excluding any and all Non-Exclusively Licensed Mundipharma Patent Claims) and Mundipharma’s interest in Joint Technology, solely to develop, register, use, sell, have sold, offer for sale and import Compound and Product in the Cidara Territory; and
(ii) a non‑exclusive, royalty‑free, fully‑paid, irrevocable, perpetual license, with the right to sublicense through multiple tiers of sublicense, under the Non‑Exclusively Licensed Mundipharma Patent Claims, solely to develop, register, use, sell, have sold, offer for sale and import Compound and Product in the Cidara Territory. In addition, Mundipharma hereby grants to Cidara an exclusive option to negotiate with Mundipharma for an exclusive, royalty-bearing license under Non‑Exclusively Licensed Mundipharma Patent Claims, solely to develop, register, use, sell, have sold, offer for sale and import Compound and Product in the Cidara Territory. For clarity, however, no right, license or option under any Non‑Exclusively Licensed Mundipharma Patent Claim is granted to Cidara or its Affiliates to develop, register, use, sell, have sold, offer for sale or import any compound that is not a Compound or any product that is not a Product, and Mundipharma hereby expressly reserves exclusively to itself all rights under the Non‑Exclusively Licensed Mundipharma Patent Claims to develop, register, use, sell, have sold, offer for sale and import all compounds that are not Compounds and all products that are not Products.
(b) Mundipharma Territory – Products Other Than Licensed Products. Subject to the terms and conditions of this Agreement (including, for clarity, Sections 4.4 and 4.5), Mundipharma hereby grants to Cidara:
(i) an exclusive (even as to Mundipharma), royalty‑free, fully‑paid, irrevocable, perpetual license, with the right to sublicense through multiple tiers of sublicense, under the Mundipharma Patents (excluding any and all Non-Exclusively Licensed Mundipharma Patent Claims), the Mundipharma Know-How (excluding Mundipharma Marketing Information), and Mundipharma’s interest in Joint Technology, in each case, solely to develop, register, use, sell, have sold, offer for sale and import Compound (other than for incorporation into Licensed Product) and Product (other than Licensed Product) in the Mundipharma Territory; and
(ii) a non‑exclusive, royalty‑free, fully‑paid, irrevocable, perpetual license, with the right to sublicense through multiple tiers of sublicense, under the Non-Exclusively Licensed Mundipharma Patent Claims, solely to develop, register, use, sell, have sold, offer for sale and import Compound (other than for incorporation into Licensed Product) and Product (other than Licensed Product) in the Mundipharma Territory. In addition, Mundipharma hereby grants to Cidara an exclusive option to negotiate with Mundipharma for an exclusive, royalty-bearing license under Non‑Exclusively Licensed Mundipharma Patent Claims, solely to develop, register, use, sell, have sold, offer for sale and import Compound (other than for incorporation into Licensed Product) and Product (other than Licensed Product) in the Mundipharma Territory.
(c) Global Development Plan and Other Development Activities for Licensed Product. Subject to the terms and conditions of this Agreement, Mundipharma hereby grants to Cidara a co‑exclusive (with Mundipharma), worldwide, royalty-free, fully-paid license, without the right to sublicense but, for clarity, with the right to subcontract, under the Mundipharma Technology and Mundipharma’s interest in Joint Technology, solely to perform development activities related to the Licensed Product, including Global Development Plan activities, including the right to file and maintain INDs for, Compound and Licensed Product in the Mundipharma Territory for such purposes.
(d) Manufacturing. Subject to the terms and conditions of this Agreement (including, for clarity, Section 2.7), Mundipharma hereby grants to Cidara:
(i) a co-exclusive (with Mundipharma), worldwide, royalty‑free, fully‑paid, irrevocable, perpetual license, with the right to sublicense through multiple tiers of sublicense, under the Mundipharma Technology (excluding any and all Non-Exclusively Licensed Mundipharma Patent Claims) and Mundipharma’s interest in Joint Technology, to make and have made Compound and Product; and
(ii) a non-exclusive, worldwide, royalty‑free, fully‑paid, irrevocable, perpetual license, with the right to sublicense through multiple tiers of sublicense, under the Non-Exclusively Licensed Mundipharma Patent Claims, solely to make and have made Compound and Product. In addition, Mundipharma hereby grants to Cidara an exclusive option to negotiate with Mundipharma for an exclusive, worldwide, royalty-bearing license under Non‑Exclusively Licensed Mundipharma Patent Claims, solely to make and have made Compound and Product. For clarity, however, no right, license or option under any Non‑Exclusively Licensed Mundipharma Patent Claim is granted to Cidara or its Affiliates to make or have made any compound that is not a Compound or any product that is not a Product, and Mundipharma hereby expressly reserves exclusively to itself all rights under the Non‑Exclusively Licensed Mundipharma Patent Claims to make and have made all compounds that are not Compounds and all products that are not Products.
Notwithstanding the co‑exclusivity of the license granted pursuant to Section 2.6(d)(i), Mundipharma retains the right to grant licenses under, and the right to subcontract to a CMO, the Mundipharma Technology and Mundipharma’s interest in Joint Technology to its Affiliates and permitted Sublicensees that have the right to market, promote and sell Licensed Product, in each case, to make and have made Compound solely for incorporation into Licensed Product, and to make and have made Licensed Product; in each case, anywhere in the world, solely for Mundipharma to develop, register, use, sell, have sold, offer for sale and import such Licensed Products in the Field in the Mundipharma Territory subject to the terms and conditions of this Agreement.
2.7 Cidara Negative Covenants. Cidara hereby covenants on behalf of itself and its Affiliates not to practice, and not to permit or cause any Affiliate of Cidara or any licensee, marketing partner or distributor of Cidara or any other Third Party to practice, any Mundipharma Technology for any purpose other than as expressly authorized in this Agreement. Cidara further covenants on behalf of itself and its Affiliates:
(a) not to develop, register, use, or seek Regulatory Approval for, Licensed Product (or Compound incorporated or for incorporation in Licensed Product) in the Mundipharma Territory; in each case, except as necessary (i) to perform Global Development Plan activities in the Mundipharma Territory, including the right to file and maintain INDs for, Compound and Licensed Product for the purpose of performing Global Development Plan activities in the Mundipharma Territory, and (ii) to perform Cidara’s obligations under Section 4.10 (CMC Development Activities);
(b) not to sell, have sold, offer for sale, or import for sale (other than to Mundipharma, its Affiliates and Sublicensees) Licensed Product (or Compound incorporated or for incorporation in Licensed Product) in the Mundipharma Territory. Without limiting the generality of the foregoing, Cidara hereby covenants on behalf of itself and Cidara-Controlled Affiliates not to, and not to permit or cause any Affiliate of Cidara to, sell or provide Compound or Licensed Product to any Third Party licensee, marketing partner or distributor of Cidara or other Third Party if any of them knows that Compound or Licensed Product sold or provided to such Third Party would be sold or transferred, directly or indirectly, for use in the Mundipharma Territory in the conduct of any activity in which Cidara itself is not permitted to engage under this Agreement. For clarity, and except as otherwise mutually agreed by the parties in writing pursuant to Section 4.3, Cidara covenants on behalf of itself and its Affiliates not to register, sell, have sold, offer for sale or import or seek Regulatory Approval for (i) any Licensed Product containing any Compound other than the Lead Compound, (ii) any Licensed Product containing the Lead Compound other than the Lead Indication Product and the Pediatric Licensed Product; or (iii) any Combination Product; in each case, in the Mundipharma Territory;
(c) except to the extent permitted by Sections 4.4 and 4.5, not to register, sell, have sold, offer for sale or import for sale, or seek Regulatory Approval for, a Product that is not Licensed Product in the Mundipharma Territory;
(d) not to conduct or have conducted any GLP Study or clinical trial of Compound or Product, except in accordance with the Global Development Plan or a protocol approved in accordance with Article 3 and the JSC Dispute Resolution Matrix; and
(e) not to grant, or purport to grant, any Affiliate of Cidara or any Third Party any license or other right to do any of the foregoing.
2.8 No Implied Licenses. No right or license under any Patents, Inventions, Data or Information of either party or its Affiliates is granted or shall be granted by implication. All such rights or licenses are or shall be granted only as expressly provided in this Agreement.
3. | GOVERNANCE |
3.1 Joint Steering Committee.
(a) Establishment and Composition. Within 30 days after the Effective Date, the parties shall establish a Joint Steering Committee (the “JSC”) composed of three representatives of each of Cidara and Mundipharma, and each party shall designate its initial JSC representatives by written notice to the other party. Each party shall be free to change its JSC representatives on written notice to the other party, provided that each party shall ensure that, at all times during the existence of the JSC, its representatives on the JSC have (i) appropriate expertise for the then-current stage of development or commercialization of Licensed Product in the Field in the Mundipharma Territory; and (ii) the authority to bind such party with respect to matters requiring approval by the JSC.
(b) Responsibilities and Authority. The JSC’s overall responsibility shall be to encourage and facilitate the exchange of information between the parties contemplated by this Agreement, and to facilitate, coordinate and oversee the development, registration and commercialization of Licensed Product in the Field in the Mundipharma Territory. The specific responsibilities of the JSC shall be:
(i) to seek harmonization in global development, regulatory approval and CMC efforts, and in relation to drug-safety matters, with respect to Licensed Product in the Field;
(ii) to serve as the principal means by which the parties review, discuss, and, to the extent required by the JSC Dispute Resolution Matrix and in accordance with Section 3.1(f), approve, amendments or updates of the Global Development Plan in accordance with Section 4.1(b);
(iii) to serve as the principal means by which the parties review, discuss, and, to the extent required by the JSC Dispute Resolution Matrix and in accordance with Section 3.1(f), approve the Mundipharma Territory Plan or any amendment thereto in accordance with Section 4.2;
(iv) to serve as the principal means by which the parties review, discuss and, to the extent required by the JSC Dispute Resolution Matrix and in accordance with Section 3.1(f), approve amendments or updates of the CMC Development Plan in accordance with Section 4.10;
(v) at the request of either party, to consider and discuss any potential Licensed Product Global Development Expansion as required by Section 4.3(a);
(vi) to review and approve the protocol (and amendments or updates thereof) for each GLP Study and clinical trial of: (A) Licensed Product proposed to be conducted by or on behalf of either party or any of its Affiliates, licensees or Sublicensees; (B) subject to Section 4.4(b), Subcutaneous Product proposed to be conducted by or on behalf of Cidara (or, after execution of a Subcutaneous Product Amendment, by or on behalf of Mundipharma) or any of its Affiliates, licensees or Sublicensees; and (C) subject to Section 4.4(b), Other Product proposed to be conducted by or on behalf of Cidara (or, after execution of an Other Product Amendment by or on behalf of Mundipharma) or any of its Affiliates, licensees or Sublicensees;
(vii) to (A) discuss any anticipated circumstance brought to the JSC’s attention by Cidara pursuant to Section 4.1(c) that could impair Cidara’s ability to perform the Global Development Plan, (B) consider measures to avoid or minimize any potential disruption to the performance of the Global Development Plan as a result thereof, and (C) approve a plan of action to implement any such measure(s) that the JSC determines to be advisable; in each case, in accordance with Section 4.1(c);
(viii) to facilitate the exchange of Cidara Know-How and Mundipharma Know-How subject to and in accordance with Section 2.3;
(ix) to serve as the principal means by which (A) Mundipharma keeps Cidara reasonably informed regarding Mundipharma’s development, registration and commercialization of Licensed Product in the Field in the Mundipharma Territory, and (B) Cidara keeps Mundipharma reasonably informed regarding Cidara’s development, registration and commercialization of Licensed Product in the Field in the Cidara Territory;
(x) to coordinate the parties’ activities in relation to international meetings and congresses that will be attended by representatives of both parties;
(xi) to review, discuss and approve opportunities to support investigator-initiated trials of, and (subject to Section 4.17) named patient programs for, the Licensed Product in the Field and to approve the supply by either party of Licensed Product for, the provision by either party of any other support of, and any other participation by either party in, any such investigator-initiated trial or such named patient program;
(xii) to establish Subcommittees (in addition to the JDC and JCC) as it deems necessary or advisable to further the purposes of this Agreement, to delegate to such Subcommittees such of the JSC’s responsibilities (excluding its responsibilities under Sections 3.1(b)(xii) and 3.1(b)(xiii)) as the JSC deems appropriate, subject to Sections 3.1(f) and 3.3, and to dissolve Subcommittees;
(xiii) to serve as a point of escalation from, and to seek to resolve disagreements and issues referred to it by, any Subcommittee; and
(xiv) to carry out such other obligations as are expressly delegated to it under this Agreement.
(c) Meetings. The JSC shall meet at least once every calendar quarter from the Effective Date until the granting of Regulatory Approval for the Lead Indication Product in the Lead Indications in the Major Markets and thereafter at such frequency as mutually agreed by the parties. JSC meetings may be conducted in person at times and places to be determined by the JSC members. Alternatively, the JSC may meet by means of teleconference, videoconference or other similar communications equipment. A reasonable number of additional representatives of a party may attend meetings of the JSC in a non‑voting capacity. Each party shall bear its own expenses of participating in meetings of the JSC. Responsibility for chairing JSC meetings shall alternate between the parties. The chair for any JSC meeting shall not have any greater authority than any other representative of either party on the JSC.
(d) Minutes. The Alliance Manager of the party responsible for chairing a JSC meeting shall be responsible for preparing the agenda and subsequent minutes of each meeting. Such Alliance Manager shall circulate a draft agenda 10 Business Days prior to the scheduled date of the JSC meeting. Such agenda shall be finalized no later than 5 Business Days prior to the scheduled JSC meeting and shall indicate which, if any, items on the agenda are Key Matters. After the JSC meeting, such Alliance Manager shall circulate a draft of the minutes of such meeting to all members of the JSC for comments within 10 Business Days after such meeting. Such minutes shall provide a description, in reasonable detail, of the discussions at the meeting and shall document all actions and determinations approved by the JSC at such meeting, as well as any Material Impact Objection raised by either party at such meeting. In addition, in the event of planned discussion at any JSC meeting of any of the matters described in Section 3.1(b)(ii), 3.1(b)(iii), 3.1(b)(iv), 3.1(b)(vi) or 3.1(b)(xi), all applicable materials, including draft plans, amendments, updates, protocols, and protocol amendments, shall be attached to the agenda as an exhibit, and, in the event of any approval by the JSC at any JSC meeting of any such plan, amendment, update, protocol or protocol amendment, such approved plan, amendment, update, protocol or protocol amendment shall be attached in final form as an exhibit to the finalized minutes of such JSC meeting. The parties shall promptly review and comment on draft minutes, and the applicable Alliance Manager shall update such draft minutes accordingly and recirculate until the parties have agreed on the final form. The parties shall cooperate in good faith to finalize the minutes no later than the date of the next JSC meeting or within 30 days of the first circulation of the draft minutes, whichever is earlier.
(e) Decision-Making. Decisions within the scope of the JSC’s authority shall be made by unanimous vote, with each party’s representatives on the JSC collectively having one
vote. The presence of at least one of each party’s JSC representatives constitutes a quorum for the conduct of business at any JSC meeting, and no vote of the JSC may be taken without a quorum present. If the JSC is unable to decide or resolve unanimously any matter within the scope of its authority set forth in Section 3.1(b), the status quo shall continue, except in the case of Key Matters, which shall be resolved in accordance with Section 3.1(f). Without limiting the generality of the foregoing, the matters described in Section 3.1(b)(xi) regarding investigator-initiated trials and named patient support shall always require unanimous approval of the JSC without resort to the dispute resolution provisions set forth in Section 3.1(f).
(f) JSC Dispute Resolution Matrix for Key Matters. If the JSC is unable to decide or resolve unanimously in accordance with Section 3.1(e) any of the matters specified in the JSC Dispute Resolution Matrix (each, a “Key Matter”), such Key Matter shall be subject to dispute resolution as specified in the JSC Dispute Resolution Matrix and as more fully described below in this Section 3.1(f).
(i) Exercise of Party’s Final Decision-Making Authority. With respect to any Key Matter that the JSC Dispute Resolution Matrix specifies is subject to final decision-making authority of a party’s JSC representatives, such party’s JSC representatives, in the exercise of their final decision-making authority, shall give good faith consideration to, and take reasonable account of, the position of the other party’s JSC representatives on such matter.
(ii) Escalation to Senior Executives. Any Key Matter that the JSC Dispute Resolution Matrix specifies is subject to Escalation to Senior Executives shall be referred to the Chief Executive Officer of Cidara and the President and CEO of Mundipharma, Europe, (in each case, such party’s “Senior Executive”) who shall promptly meet and attempt in good faith to resolve such issue within 30 days.
(iii) Material Impact Objection. If a party proposes a Key Matter specified in the JSC Dispute Resolution Matrix in respect of which the other party has the right to raise a Material Impact Objection (defined below), and such other party in good faith believes that the proposed Key Matter is substantially likely to have a Material Impact on the Licensed Product, then such other party’s JSC representatives may instruct the Alliance Manager with responsibility for preparing the minutes of the applicable JSC meeting to reflect in such minutes that it objects to such proposed Key Matter on the basis of such Material Impact (such objection, a “Material Impact Objection”). Alternatively, a party may raise a Material Impact Objection either before the JSC meeting at which a Key Matter proposed by the other party is to be discussed or within 15 Business Days after such JSC meeting by delivering notice (including by email) to the proposing party’s Alliance Manager, who shall promptly bring the matter to the attention of the JSC, and if such Material Impact Objection is raised after such JSC meeting, the JSC shall promptly convene a meeting to discuss such Material Impact Objection. As specified in the JSC Dispute Resolution Matrix, any such Key Matter with respect to which a party raises a Material Impact Objection that is not resolved by unanimous vote of the JSC shall first be escalated to the Senior Executives for attempted resolution in accordance with Section 3.1(f)(ii) and, if the Senior Executives are unable to resolve such Key Matter, shall be […***…], and the party proposing such Key Matter shall not proceed with the activity or course of action that is the subject of such Material Impact Objection unless and until such Material Impact Objection is resolved in
such party’s favor in accordance with the foregoing procedures and pursuant to and in accordance with Section 3.1(f)(iv). If the JSC or the Senior Executives agree, […***…] that the applicable activity or course of action does not pose a substantial risk of a Material Impact […***…], the parties shall be bound by such determination […***…]. If the JSC or the Senior Executives agree, […***…] that the applicable activity or course of action should not be undertaken because it poses a substantial risk of a Material Impact, the parties shall be bound by such determination, and the proposing party shall not have the right to conduct such activity or take such course of action […***…].
(iv) […***…] Any Key Matter specified in the JSC Dispute Resolution Matrix with respect to which a party has a right to and does raise a Material Impact Objection that is not resolved by the Senior Executives in accordance with Section 3.1(f)(ii) shall be […***…]. Promptly after […***…]. The parties shall mutually agree on the medium by which each party may present its position […***…] (i.e., in writing and/or orally) and shall establish a reasonable limit on the length (i.e., page limit and/or duration) of each party’s presentation. Each party may, but is not required to, include in its presentation a recommendation that […***…]. The parties shall share equally the fees and costs of […***…], regardless of the […***…] ultimate determination. The questions to be posed to […***…] are:
(A) | […***…] |
(B) | […***…] |
(C) | […***…] |
[…***…]
If the answer to the questions in (A) and (B) are both “Yes”, the proposed activity or course of action […***…]. The party proposing the applicable activity or course of action shall consider in good faith[…***…].
If the answer to the question in (A) is “No” and the answer to the question in (C) is “Yes”, the proposed activity or course of action […***…]. The party proposing the applicable activity or course of action shall consider in good faith[…***…]
(v) No Arbitration. The parties intend that all matters within the scope of the JSC’s decision-making authority shall be resolved by the parties in accordance with Section 3.1(e), this Section 3.1(f) and the JSC Dispute Resolution Matrix, and no matter within the scope of the JSC’s authority shall be subject to the dispute resolution mechanisms set forth in Article 12.
3.2 Subcommittees.
(a) Within 30 days after the Effective Date, the parties shall establish a Joint Development Committee (the “Joint Development Committee” or “JDC”) to oversee activities needed to obtain Regulatory Approvals for Licensed Product in the Field in the Mundipharma Territory.
(b) Within 30 days after the Effective Date, the parties shall establish a Joint Manufacturing Committee (the “Joint Manufacturing Committee” or “JMC”) to oversee activities in relation to the manufacturing of Compound and Licensed Product, including CMC development activities to generate Data needed to obtain Regulatory Approvals for Licensed Product in the Field in the Mundipharma Territory, and in relation to the CMC Development Plan.
(c) As soon as reasonably practical but no later than 12 months prior to the anticipated date of Regulatory Approval of the Lead Indication Product in the Treatment Indication in the Major European Countries, the parties shall establish a Joint Commercialization Committee (the “Joint Commercialization Committee” or “JCC”) to oversee commercialization of Licensed Product in the Field in the Mundipharma Territory.
(d) In addition, from time to time, the JSC may establish additional subcommittees to which it may delegate specified projects or activities within the scope of authority of the JSC, as it deems necessary or advisable. Each Subcommittee shall be composed of an equal number of representatives of each party as the JSC determines is appropriate from time to time and shall meet with such frequency as the applicable Subcommittee shall determine.
Matters relating to the composition (other than the number of representatives), meetings and minutes of each Subcommittee shall be as set forth in Sections 3.1(a) through 3.1(e), mutatis mutandis. Decisions within the scope of the Subcommittee’s authority shall be made by unanimous vote, with each party’s representatives on the Subcommittee collectively having one vote. The presence of at least one of each party’s Subcommittee representatives constitutes a quorum for the conduct of business at any Subcommittee meeting, and no vote of the Subcommittee may be taken without a quorum present. If, with respect to a matter that is subject to a Subcommittee’s decision-making authority, the Subcommittee cannot reach unanimity, the matter shall be referred to the JSC for resolution in accordance with Section 3.1(f) and the JSC Dispute Resolution Matrix.
3.3 Scope of Authority; Exclusions. Notwithstanding the establishment and existence of the JSC or any Subcommittee, each party shall retain the rights, powers and discretion granted to it hereunder, and neither the JSC nor any Subcommittee shall be delegated or vested with rights, powers or discretion unless such delegation or vesting is expressly provided herein. In addition to any other exclusions from or limitations on its authority set forth in this Article 3 or elsewhere in this Agreement, the JSC shall have no right or authority:
(a) to interpret, modify, amend, or waive compliance with any provision of, or any right or remedy under, this Agreement;
(b) to approve any Licensed Product Global Development Expansion (each of which shall require the negotiation and execution by the parties of an Expanded Licensed Product Amendment as described in Section 4.3(a) or Section 4.3(c);
(c) to approve a Subcutaneous License Expansion (which shall require the negotiation and execution by the parties of a Subcutaneous Product Amendment as described in Section 4.4);
(d) to approve the expansion of the scope of the licenses granted under this Agreement to include an Other Product (which shall require the negotiation and execution by the parties of an Other Product Amendment for such Other Product as described in Section 4.5);
(e) to determine whether or not a party has complied with any of its obligations under this Agreement;
(f) to determine whether or not, or when, any milestone event set forth in Section 5.3, 5.5 or 5.6 has been achieved;
(g) to determine any issue in a manner that would conflict with the express terms of this Agreement; or
(h) to make any decision or approve any matter that is expressly stated to require the mutual written agreement of the parties or the written consent of one or both parties.
3.4 Alliance Managers. Within 30 days after the Effective Date, each party shall appoint (and notify the other party of the identity of) a representative of such party to act as the primary point of contact for the parties with responsibility for the smooth functioning of the alliance by creating and maintaining collaborative, efficient, and responsive communications within and between Mundipharma and Cidara (each, an “Alliance Manager”). A party may replace its Alliance Manager on written notice to the other party and may designate a substitute to temporarily perform the functions of that Alliance Manager by written notice to the other party. The Alliance Managers shall support alliance governance activities and shall attend all JSC meetings (except as otherwise determined by the JSC) and support their chairperson of the JSC in the discharge of their responsibilities but shall be non-voting participants in such JSC meetings. The Alliance Managers shall also: (i) support project teams across all functions in meeting the objectives for this Agreement; (ii) take responsibility for ensuring that governance activities, such as the conduct of required JSC meetings and production of meeting agendas and minutes occur as set forth in this Agreement, and that relevant action items resulting from such meetings are appropriately carried out or otherwise addressed; (iii) bring to the attention of the JSC (and other Subcommittees in a timely manner any matters or issues they reasonably believe should be discussed; (iv) serve as the primary point of communication for seeking consensus between the parties regarding issues; and (v) attend meetings of the JDC, JCC or other Subcommittee as may be deemed appropriate.
4. | DEVELOPMENT AND REGULATORY |
4.1 Global Development Program and Development in the Cidara Territory.
(a) Global Development Plan. Subject to the remainder of this Section 4.1(a) and to Section 5.2, Cidara shall use Commercially Reasonable Efforts to conduct, or have conducted, the Global Development Plan in accordance with the budgeted costs and timelines. Upon Cidara’s reasonable request, Mundipharma shall provide reasonable cooperation and informal, non-financial (other than the Global Development Costs) assistance to Cidara in connection with (i) Cidara’s management and execution of the Lead Indication Trial activities that are conducted in the Mundipharma Territory, (ii) Cidara’s filing of INDs with Regulatory Authorities in the Mundipharma Territory for the Lead Indication Trials and in interactions with such Regulatory Authorities in connection therewith, and (iii) Cidara’s management and execution of the CMC development activities set forth in the Global Development Plan. Each party shall keep the JSC and JDC regularly informed of the status, progress and results of all Global Development Plan activities conducted by it or on its behalf.
(b) Amendments to Global Development Plan. The parties shall periodically review and, if necessary, propose updates to the then-current Global Development Plan for discussion at the JSC; provided that Cidara shall be responsible for preparing any formal written amendment to the Global Development Plan for review, and, if applicable, approval, by the JSC. The parties shall discuss in good faith, and attempt to reach consensus regarding, any such proposed modification to the Global Development Plan. Approval requirements for amendments or updates to the Global Development Plan shall be as set forth in Sections 3.1(b)(ii), 3.1(b)(vi) and 3.1(e), and, if applicable, Section 3.1(f) and the JSC Dispute Resolution Matrix. References to the “Global Development Plan” in this Agreement shall be construed to refer to the Global Development Plan, as then in effect (including all updates and amendments thereto made in accordance with Sections 3.1(b)(ii), 3.1(b)(vi) and 3.1(e), and, if applicable, Section 3.1(f) and the JSC Dispute Resolution Matrix.
(c) Anticipated Performance Issue. If, during the performance of the Global Development Plan, Cidara anticipates that any change in Cidara’s circumstances occurring after the Effective Date is likely to arise that would impair Cidara’s ability to perform the Global Development Plan, (i) Cidara’s Alliance Manager shall promptly inform Mundipharma’s Alliance Manager thereof, (ii) each party’s Alliance Manager shall promptly bring such anticipated change in circumstances to the attention of such party’s JSC representatives, and a meeting of the JSC shall be convened as promptly as practicable to discuss the matter. At such meeting, the parties’ JSC representatives shall discuss such anticipated change in circumstances, consider practical measures that may be taken to avoid or minimize any potential disruption to the performance of the Global Development Plan, and cooperate in good faith to formulate a plan of action to implement any such measure(s) that the JSC determines to be advisable. By way of example and not of limitation, such measures could include Mundipharma’s assumption of responsibility for overseeing or performing one or more Global Development Plan activities, or Mundipharma making payments on behalf of Cidara to Third Party contractors performing Global Development Plan activities. If the parties agree to Mundipharma making payments on behalf of Cidara to such Third Parties, (i) Mundipharma shall have the right to deduct the payments from future payment obligations of Mundipharma to Cidara arising under this Agreement, and (ii) any activity under the Global Development Plan for which Mundipharma makes any such payment on behalf of Cidara in an amount that exceeds the greater of (x) […***…]% of the total cost of such activity and (y) $[…***…], shall be deemed to be Key Matters pursuant to Section 3.1(f) […***…].
(d) Development in the Cidara Territory. Cidara shall in a timely manner keep the JSC and JDC reasonably and regularly informed of the status, progress and results of any GLP Study and clinical trial of Licensed Product in the Field in the Cidara Territory and in the Mundipharma Territory that are not part of the Global Development Plan.
4.2 Mundipharma Territory Plan.
(a) Mundipharma shall be solely responsible for conducting, at Mundipharma’s sole expense, such Mundipharma Territory-Specific development activities with respect to Lead Indication Product in the Lead Indications and a Pediatric Licensed Product in the Mundipharma Territory as may be required by applicable Regulatory Authorities to support MAA filing and Regulatory Approval in the Mundipharma Territory, including the European Paediatric Investigational Plan. Upon Mundipharma’s reasonable request, Cidara shall provide reasonable cooperation and informal, non-financial assistance to Mundipharma in connection with the Mundipharma Territory Plan.
(b) Mundipharma shall have the right to amend or update the Mundipharma Territory Plan from time to time, subject to Sections 3.1(b)(iii), 3.1(b)(vi) and 3.1(e) and, if applicable, Section 3.1(f) and the JSC Dispute Resolution Matrix, provided that at all times the activities covered by the Mundipharma Territory Plan shall be limited to Mundipharma Territory-Specific development activities with respect to Lead Indication Product in the Lead Indications. References to the “Mundipharma Territory Plan” in this Agreement shall be construed to refer to the Mundipharma Territory Plan, as then in effect (including all amendments thereto).
(c) Mundipharma shall in a timely manner keep the JSC and JDC reasonably and regularly informed of the status, progress and results of all Mundipharma Territory Plan activities.
(d) Mundipharma shall be responsible for conducting the European Paediatric Investigational Plan with respect to a Pediatric Licensed Product as required by the EMA to maintain the Regulatory Approval for the Lead Indication Product.
4.3 Expanded Licensed Products.
(a) Licensed Product Global Development Expansion. The parties acknowledge that the scope of the Global Development Plan and the Mundipharma Territory Plan as of the Effective Date is limited to development of Lead Indication Product for the Lead Indications and the Pediatric Licensed Product. However, the parties also acknowledge that it may be beneficial to both parties to pursue development of an Expanded Licensed Product. At the request of either party from time to time, the JSC shall consider and discuss the potential expansion of the scope of the parties’ collaborative development efforts under this Agreement to encompass development of an Expanded Licensed Product (on an New Indication-by-New Indication and/or Expanded Licensed Product-by-Expanded Licensed Product (as applicable) basis, a “Licensed Product Global Development Expansion”). For clarity, (A) Cidara shall be free to conduct development of any Expanded Licensed Product, provided that the protocol for any GLP Study or clinical trial of such Expanded Licensed Product shall require JSC approval under Section 3.1(b)(vi), subject, if applicable, to Section 3.1(f) and the JSC Dispute Resolution Matrix; and (B) Mundipharma shall not have the right to conduct any GLP Study or clinical trial of any Expanded Licensed Product, without first (x) obtaining required approval of the protocol for any such GLP Study or clinical trial of any of the foregoing under Sections 3.1(b)(vi) and, if applicable, to Section 3.1(f) and the JSC Dispute Resolution Matrix, and (y) proposing a Licensed Product Global Development Expansion for the same to the JSC for consideration and discussion as set forth above. (For clarity, the parties acknowledge and agree that the Pediatric Licensed Product shall not require any Licensed Product Global Development Expansion and that Mundipharma shall be free to conduct the European Pediatric Investigational Plan without following the procedures set forth in this Section 4.3, subject to obtaining required approval of the protocol for the European Pediatric Investigational Plan under Sections 3.1(b)(vi) and, if applicable, to Section 3.1(f) and the JSC Dispute Resolution Matrix.) If the parties reach consensus at the JSC level to pursue a Licensed Product Global Development Expansion for an Expanded Licensed Product, such expansion shall become effective only upon:
(i) mutual written agreement of the parties on: (A) a mutually-agreed forecast of the peak incremental Net Sales of such Expanded Licensed Product in the Mundipharma Territory over (x) […***…] (for each such Expanded Licensed Product, the “Mundipharma Peak Incremental Net Sales Forecast”); and (B) a mutually-agreed forecast of the peak incremental Net Sales of such Expanded Licensed Product in the Cidara Territory over […***…] (for each Expanded Licensed Product, the “Cidara Peak Incremental Net Sales Forecast” and, collectively with the Mundipharma Peak Incremental Net Sales Forecast for such Expanded Licensed Product, the “Peak Incremental Net Sales Forecasts”). In the event that the parties are unable to reach mutual agreement regarding the Peak Incremental Net Sales Forecasts for such Expanded Licensed Product, then upon the request of either party, the matter shall be referred to the Senior Executives, who shall promptly meet and attempt in good faith to agree upon such Peak Incremental Net Sales Forecasts within 30 days. If the Senior Executives are unable to agree upon such Peak Incremental Net Sales Forecasts within such 30-day period, then the Peak Incremental Net Sales Forecasts shall be determined in accordance with Section 12.5; and
(ii) execution by the parties of a written amendment to this Agreement (each, a “Expanded Licensed Product Amendment”) under which:
(1) an initial Expanded Licensed Product Global Development Plan and an initial Expanded Licensed Product Mundipharma Territory Plan for such Expanded Licensed Product would be attached as exhibits;
(2) the parties would share all Global Development Costs (mutatis mutandis) incurred after the execution of such Expanded Licensed Product Amendment in performing the Expanded Licensed Product Global Development Plan activities for such Expanded Licensed Product (for each Expanded Licensed Product, “Global Development Expansion Costs”), in accordance with Section 6.1(a), mutatis mutandis, provided that […***…] shall, unless otherwise mutually agreed by the parties in writing, be determined by […***…];
(3) with respect only to an Expanded Licensed Product referred to in clause (A) of Section 1.47 (New Indications) or in clause (C) of Section 1.47 (Combination Products), Mundipharma would pay an additional one-time, non‑refundable, non‑creditable Global Development Expansion fee to Cidara equal to […***…]% of the Mundipharma Peak Incremental Net Sales Forecast for such Expanded Licensed Product (such Global Development Expansion Fee not to exceed $[…***…]) in recognition of the approval of a MA Variation for the Expanded Licensed Product (New Indications) or approval of a MAA for the Expanded Product (Combination Product) in […***…] (the “Global Development Expansion Fee”), which shall be payable as follows:
(A) Mundipharma would pay 100% of the Global Development Expansion Fee within 10 Business Days of the first approval of a MA Variation or of a MAA, as applicable, for the Expanded Licensed Product in […***…]; or
(B) if the approval of a MA Variation or of a MAA approval, as applicable, for the Expanded Licensed Product in […***…] occurs before the approval of a corresponding MA Variation, or of a corresponding MAA, as applicable, for the Expanded Licensed Product in […***…],
(C)
Mundipharma would pay […***…]% of the Global Development Expansion Fee within 10 Business Days of the approval of the MA Variation or MAA, as applicable, for the Expanded Licensed Product in […***…] (i.e., […***…]% of the agreed Peak Incremental Net Sales Forecast for such Expanded Licensed Product) and, if applicable, the remaining […***…]% of the Global Development Expansion Fee within 10 Business Days of the approval of a MA Variation or MAA, as applicable, for the Expanded Licensed Product in […***…] (i.e., the remaining […***…]% of the agreed Peak Incremental Net Sales Forecast for such Expanded Licensed Product);
(4) Net Sales of all Licensed Products (including Lead Indication Product and Pediatric Licensed Product) for all indications in the Mundipharma Territory would be aggregated for purposes of the calculation of royalties under Section 5.7 and determination of the achievement of Commercial Milestone Events under Section 5.6; and
(5) except as otherwise set forth in this Section 4.3(a)(ii), Mundipharma would not have any other obligations to pay Cidara any additional amounts pursuant to the Expanded Licensed Product Amendment.
If the parties reach consensus at the JSC level to pursue a Licensed Product Global Development Expansion for an Expanded Licensed Product, the parties shall negotiate in good faith with the objective of entering into an Expanded Licensed Product Amendment as described above in an expeditious manner. Any Expanded Licensed Product Amendment would contain other appropriate and commercially reasonable terms, provided that, except as otherwise expressly set forth in the preceding subparagraphs (1) through (5) of this Section 4.3(a)(ii), the general expectation of the parties is that the terms and conditions of this Agreement would apply to such Licensed Product Global Development Expansion to the same extent as they apply to the Lead Indication Product for the Lead Indications and to the Pediatric Licensed Product. For clarity, neither party shall be obligated to enter into any Expanded Licensed Product Amendment.
(b) Election Not to Pursue Licensed Product Global Development Expansion. If a party proposes at the JSC level a Licensed Product Global Development Expansion for an Expanded Licensed Product, but either (i) the other party does not wish to pursue such Licensed Product Global Development Expansion for such Expanded Licensed Product or (ii) the parties reach consensus at the JSC level to pursue a Licensed Product Global Development Expansion for such Expanded Licensed Product but are unable to conclude an Expanded Licensed Product Amendment for such Licensed Product Global Development Expansion within […***…] after initiating good faith negotiations regarding such expansion, then, in each case, a party desiring to develop such Expanded Licensed Product shall be free to conduct independent development of such Expanded Licensed Product, subject to Section 3.1(b)(vi) and, if applicable, Section 3.1(f) and the JSC Dispute Resolution Matrix, and to register and commercialise such Expanded Licensed Product solely in its respective Territory. In such event, the other party shall not be entitled to use any Expanded Licensed Product Clinical Efficacy Data generated by or on behalf of the first party in the development of such Expanded Licensed Product in support of any NDA, MAA, MA Variation or other Product Filing or for obtaining or maintaining Regulatory Approval for such Expanded Licensed Product in the other party’s Territory or in connection with the commercialization of such Expanded Licensed Product in the other party’s Territory, except as expressly set forth in Section 4.3(c).
(c) One-Time Buy-In Right for Global Development Expansion. In the absence of a Licensed Product Global Development Expansion for an Expanded Licensed Product, if Cidara, pursuant to the first paragraph of Section 4.3(a), or either party, pursuant to Section 4.3(b), pursues independent clinical development of such Expanded Licensed Product, such party (the “Independent Development Party”) shall, within […***…] after the first availability of top-line results from the […***…], deliver such top-line results to the other party, together with a written report of the reasonable and documented external costs and expenses incurred by the Independent Development Party to date in connection with such clinical trial or preparing, submitting, obtaining or maintaining Product Filings specific to such clinical trial (the “Independent Expanded Licensed Product Costs”). In such event, the other party shall have the one-time right, exercisable solely during the 60-day period after receipt of such top-line results and the report of Independent Expanded Licensed Product Costs, to notify the Independent Development Party that the other party wishes to pursue a Licensed Product Global Development Expansion for such Expanded Licensed Product, in which event the parties shall negotiate in good faith for up to an additional 120 days (or such longer period as the parties may agree) with the objective of executing an Expanded Licensed Product Amendment for such Expanded Licensed Product in accordance with Section 4.3(a), mutatis mutandis; provided, however, that, in addition to the terms set forth in subparagraphs (1) through (4), and notwithstanding subparagraph (5), of Section 4.3(a)(ii), such Expanded Licensed Product Amendment shall: (i) obligate the other party to pay to the Independent Development Party a buy-in fee in an amount equal to […***…]% of the applicable share of the Independent Expanded Licensed Product Costs incurred by the Independent Development Party up to the date of such Expanded Licensed Product Amendment, such share being determined by […***…] (each, an “Expanded Licensed Product Buy‑In Fee”), it being understood that such total Independent Expanded Licensed Product Costs may include additional Independent Expanded Licensed Product Costs incurred after the date of the written cost report delivered to the other party as described above in this Section 4.3(c); and (ii) entitle the other party, upon payment of such Expanded Licensed Product Buy-In Fee, to use the Expanded Licensed Product Clinical Efficacy Data generated by or on behalf of the Independent Development Party with respect to such Expanded Licensed Product prior to execution of such Expanded Licensed Product Amendment in support of any IND, NDA, MAA, MA Variation, or other Product Filing or Regulatory Approval for such Expanded Licensed Product in the other party’s Territory or in connection with the commercialization of such Expanded Licensed Product in the other party’s Territory.
4.4 Subcutaneous Product Option.
(a) Subcutaneous Product Option Grant. Mundipharma acknowledges that, as of the Effective Date, the License granted to Mundipharma under this Agreement excludes any rights with respect to Subcutaneous Product (or any other Product that is not a Licensed Product). Subject to the terms and conditions of this Section 4.4, Cidara hereby grants to Mundipharma the exclusive option, exercisable only one time and solely during the Subcutaneous Product
Option Period in accordance with Section 4.4(c), to expand the License to include Subcutaneous Product (the “Subcutaneous Product Option”). During the Subcutaneous Product Option Period and, if Mundipharma exercises the Subcutaneous Product Option prior to expiration of the Subcutaneous Product Option in accordance with Section 4.4(c), during the Subcutaneous Negotiation Period, Cidara and its Affiliates shall not grant, or offer to grant, any Third Party any license under Cidara Technology (mutatis mutandis with respect to the Subcutaneous Product) or Cidara’s interest in Joint Technology (mutatis mutandis with respect to the Subcutaneous Product), to develop, register, use, sell, have sold, offer for sale or import Subcutaneous Product in the Field in the Mundipharma Territory, or any option or other right to obtain such license, or otherwise restrict or encumber Mundipharma’s rights and ability to expand the License to include Subcutaneous Product; provided, however, that the foregoing shall not be construed: (i) to prevent or restrict Cidara and its Affiliates from contracting with Third Party contract service providers to perform development activities with respect to Subcutaneous Product on behalf of Cidara or its Affiliates anywhere in the world, subject to Section 3.1(b)(vi) and, if applicable, Section 3.1(f) and the JSC Dispute Resolution Matrix; or (ii) transferring or selling all or substantially all of Cidara’s business related to Compound and Product to a Third Party, whether by merger, sale of stock, sale of assets or otherwise, in conjunction with the assignment of this Agreement and all of Cidara’s rights and obligations hereunder, to a Third Party in accordance with Section 13.7. Notwithstanding the foregoing or any other provision of this Agreement to the contrary, Cidara shall at all times be free to conduct or have conducted, and no JSC approval (or any approval by the Senior Executives or […***…]) shall be required for the conduct of, that certain Phase 1 clinical trial of Subcutaneous Product conducted with the U.S. National Institutes of Health to evaluate safety, tolerability, and pharmacokinetics for Subcutaneous Product (the “NIH Trial”).
(b) Cidara Development of Subcutaneous Product During Subcutaneous Product Option Period. During the Subcutaneous Product Option Period, Cidara may, but is not obligated to, develop Subcutaneous Product for one or more indications in the Field throughout the world, subject to Section 3.1(b)(vi) and, if applicable, Section 3.1(f) and the JSC Dispute Resolution Matrix. Cidara shall keep Mundipharma reasonably and regularly informed, primarily via the JSC or JDC, of the plans for, and the status, progress and results of, development activities with respect to Subcutaneous Product and regulatory activities related thereto. In addition, Cidara shall provide to Mundipharma (i) top-line data from any clinical trial of Subcutaneous Product that is completed during the Subcutaneous Product Option Period, promptly following the first availability thereof, (ii) true and complete copies of all final reports of any GLP Study or clinical trial of Subcutaneous Product conducted by or on behalf of Cidara or Cidara-Controlled Affiliates that are received by Cidara or Cidara-Controlled Affiliates during the Subcutaneous Product Option Period, and (iii) copies of written advice received by Cidara or Cidara-Controlled Affiliates from, and copies of Product Filings (including INDs, in each case to the extent made by Cidara or received by Cidara directly from a Regulatory Authority) with, Regulatory Authorities regarding the development and Regulatory Approval of Subcutaneous Product. No later than […***…] before […***…] in the Mundipharma Territory (in either case, the “Subcutaneous Efficacy Trial”), Cidara shall provide Mundipharma with written notice thereof and a summary of […***…] (the “Subcutaneous Efficacy Trial Notice”).
(c) Subcutaneous Product Option Exercise; Subcutaneous Product Amendment. Subject to the terms and conditions of this Agreement, Mundipharma may exercise the Subcutaneous Product Option at any time (but only one time) during the Subcutaneous Product Option Period by delivering written notice of such exercise (“Exercise Notice”) to Cidara; provided that the expansion of the License to include the Subcutaneous Product shall become effective only upon payment in full of the Subcutaneous Product Option Payment. Promptly following delivery of the Exercise Notice, the parties shall negotiate in good faith a written amendment to this Agreement (a “Subcutaneous Product Amendment”) to which an initial Subcutaneous Global Development Plan and an initial Subcutaneous Mundipharma Territory Plan would be attached as exhibits, and which would include, without limitation, the following provisions:
(i) the expansion of the License and the Grant-Back License to include the Subcutaneous Product (collectively, the “Subcutaneous License Expansion”);
(ii) the Subcutaneous License Expansion would become effective upon the payment by Mundipharma to Cidara of an option payment equal to […***…]% of the Subcutaneous Peak Incremental Net Sales Forecast, to be determined as set forth below (the “Subcutaneous Option Payment”), such Subcutaneous Option Payment not to exceed $[…***…]. For clarity, the amount of the Option Payment (in U.S. dollars) would be specified in the Subcutaneous Product Amendment. Promptly following delivery of the Exercise Notice to Cidara, the parties shall mutually agree in good faith upon: (A) a forecast of the peak incremental Net Sales (mutatis mutandis) of Subcutaneous Product in the Mundipharma Territory (the “Mundipharma Subcutaneous Peak Incremental Net Sales Forecast”); and (B) a forecast of the peak incremental Net Sales (mutatis mutandis) of Subcutaneous Product in the Cidara Territory (the “Cidara Subcutaneous Peak Incremental Net Sales Forecast” and, collectively with the Mundipharma Subcutaneous Peak Net Sales Forecast, the “Subcutaneous Peak Net Sales Forecasts”); each of which shall be determined in accordance with the same methodology, which shall be mutually agreed upon by the parties in good faith. In the event that the parties are unable to reach mutual agreement regarding the Subcutaneous Peak Incremental Net Sales Forecasts, including the methodology to be used to determine the Subcutaneous Peak Incremental Net Sales Forecasts, then upon the request of either party, the matter shall be referred to the Senior Executives, who shall promptly meet and attempt in good faith to agree upon the Subcutaneous Peak Incremental Net Sales Forecasts and such methodology within 30 days. If the Senior Executives are unable to agree upon the Subcutaneous Peak Incremental Net Sales Forecasts and such methodology within such 30-day period, then the Subcutaneous Peak Incremental Net Sales Forecasts, including the methodology to be used to determine the Subcutaneous Peak Incremental Net Sales Forecasts, shall be determined in accordance with Section 12.5;
(iii) Cidara would use Commercially Reasonable Efforts to conduct, or have conducted, those development activities with respect to Subcutaneous Product in the Subcutaneous Indication(s) as are set forth in the Subcutaneous Global Development Plan. The provisions of Articles 3 and 4 (excluding this Section 4.4) would apply, mutatis mutandis, to Subcutaneous Product;
(iv) the parties would share all Global Development Costs (mutatis mutandis) incurred after delivery of the Exercise Notice in performing Subcutaneous Global Development Plan activities for the Subcutaneous Indication(s) (“Subcutaneous Global Development Costs”), in accordance with Section 6.1(a), mutatis mutandis, provided that […***…] shall, unless otherwise mutually agreed by the parties in writing, be determined by […***…];
(v) the provisions of Article 2 would apply, mutatis mutandis, to Subcutaneous Product;
(vi) within 30 days following the first achievement (whether by Mundipharma, an Affiliate of Mundipharma or a Sublicensee) of each of the milestone events set forth in the table below, Mundipharma would provide Cidara with written notice of such achievement and pay to Cidara the corresponding non‑refundable, non‑creditable milestone payment set forth in such table, subject to Section 5.4, mutatis mutandis:
Subcutaneous Regulatory Milestone Event | Payment (USD) |
1. […***…] | $[…***…]* |
2. […***…] | $[…***…]* |
* | Aggregate amount payable for achievement in all Major European Countries. |
(vii) Net Sales of all Licensed Products (including Lead Indication Product and Pediatric Licensed Product) and Subcutaneous Product for all indications in the Mundipharma Territory would be aggregated for purposes of the calculation of royalties under Section 5.7 and determination of the achievement of Commercial Milestone Events under Section 5.6.
(viii) except as otherwise set forth in this Section 4.4(c), Mundipharma would not have any other obligations to pay Cidara or any of its Affiliates any additional amounts pursuant to the Subcutaneous Product Amendment, including any upfront payments or any development, regulatory or commercialization milestone payments.
If Mundipharma exercises the Subcutaneous Product Option as set forth above, the parties shall negotiate in good faith for up to […***…] (as such period may be extended by mutual written agreement of the parties, the “Subcutaneous Negotiation Period”) with the objective of entering into the Subcutaneous Product Amendment before expiration of the Subcutaneous Negotiation Period. The Subcutaneous Product Amendment would contain other appropriate and commercially reasonable terms, provided that, except as otherwise expressly set forth in the preceding subparagraphs (i) through (viii) of this Section 4.4(c), the general expectation of the parties is that the terms and conditions of this Agreement would apply to such Subcutaneous
License Expansion, mutatis mutandis, to the same extent as they apply to the Lead Indication Product and the Pediatric Licensed Product for the Lead Indications.
(d) Failure to Exercise Subcutaneous Product Option or to Execute Subcutaneous Product Amendment. If Mundipharma fails to deliver an Exercise Notice prior to expiration of the Subcutaneous Product Option Period, or if Mundipharma delivers an Exercise Notice prior to expiration of the Subcutaneous Product Option Period and the parties engage in negotiations of a Subcutaneous Product Amendment but are unable to conclude such Subcutaneous Product Amendment prior to expiration of the Subcutaneous Negotiation Period, then Cidara shall be free to develop, register and commercialize Subcutaneous Product throughout the world, and to grant licenses to one or more Third Parties to do so, without obligation to Mundipharma; provided, however, that if the parties had engaged in negotiations of a Subcutaneous Product Amendment but were unable to conclude such Subcutaneous Product Amendment prior to expiration of the Subcutaneous Negotiation Period, then, during the […***…] period commencing upon expiration of the Subcutaneous Negotiation Period, Cidara and its Affiliates shall not […***…].
(e) Cidara Launch of Subcutaneous Product in Mundipharma Territory. If the Subcutaneous Product is launched by Cidara, any of its Affiliates or Third Party licensees in any country within the Mundipharma Territory, then […***…], and, […***…], Net Sales of such Licensed Product in such country are […***…], […***…] in accordance with this Section 4.4(e). For purposes of this Section 4.4(e), “Independent Factor” shall mean […***…]. Subject to the preceding provisions of this Section 4.4(e), on a country-by-country basis, […***…].
The […***…] pursuant to this Section 4.4(e) shall be calculated as follows:
[…***…]
Where: (A) […***…]; (B) […***…]; (C) […***…]; and (D) […***…].
Notwithstanding the foregoing:
(x) if […***…], there shall be […***…];
(y) Mundipharma shall […***…] under this Section 4.4(e) if […***…]; and
(z) the […***…].
[…***…]
[…***…]
4.5 Option for Other Products.
(a) Other Product Option Grant. Mundipharma acknowledges that, as of the Effective Date, the License granted to Mundipharma under this Agreement excludes any rights with respect to any Product in a formulation for non-intravenous and non-subcutaneous administration – i.e., a Product that is neither a Licensed Product nor a Subcutaneous Product (on a formulation for other administration-by-formulation for other administration basis, an “Other Product”). Subject to the terms and conditions of this Section 4.5, Cidara hereby grants to Mundipharma the exclusive option, exercisable only one time per Other Product and solely during
the Other Product Option Period for each Other Product in accordance with Section 4.5(c), to expand the License to include such Other Product (each, an “Other Product Option”) on the terms and conditions of Section 4.4(c), which shall apply mutatis mutandis to such Other Product and Other Product Option, except as expressly provided in Section 4.5(c). During the Other Product Option Period for an Other Product and, if Mundipharma exercises the Other Product Option for such Other Product prior to expiration of the applicable Other Product Option in accordance with Section 4.4(c), mutatis mutandis, during the Other Product Negotiation Period, mutatis mutandis, for such Other Product, Cidara and its Affiliates shall not grant, or offer to grant, any Third Party any license under Cidara Technology (mutatis mutandis with respect to the applicable Other Product) or Cidara’s interest in Joint Technology (mutatis mutandis with respect to the applicable Other Product), to develop, register, use, sell, have sold, offer for sale or import such Other Product in the Field in the Mundipharma Territory, or any option or other right to obtain such license, or otherwise restrict or encumber Mundipharma’s rights and ability to expand the License to include such Other Product; provided, however, that the foregoing shall not be construed: (i) to prevent or restrict Cidara and its Affiliates from contracting with Third Party contract service providers to perform development activities with respect to such Other Product on behalf of Cidara or its Affiliates anywhere in the world, subject to Section 3.1(b)(vi) and, if applicable, Section 3.1(f) and the JSC Dispute Resolution Matrix; or (ii) transferring or selling all or substantially all of Cidara’s business related to Compound and Product to a Third Party, whether by merger, sale of stock, sale of assets or otherwise, in conjunction with the assignment of this Agreement and all of Cidara’s rights and obligations hereunder, to a Third Party in accordance with Section 13.7.
(b) Cidara Development of Other Product During Other Product Option Period. On an Other Product-by-Other Product basis, during the Other Product Option Period for an Other Product, Cidara may, but is not obligated to, develop such Other Product for one or more indications in the Field throughout the world, subject to Section 3.1(b)(vi) and, if applicable, Section 3.1(f) and the JSC Dispute Resolution Matrix. Cidara shall keep Mundipharma reasonably and regularly informed, primarily via the JSC or JDC, of the plans for, and the status, progress and results of, development activities with respect to such Other Product and regulatory activities related thereto. In addition, Cidara shall provide to Mundipharma (i) top-line data from any clinical trial of such Other Product that is completed during the applicable Other Product Option Period, promptly following the first availability thereof, (ii) true and complete copies of all final reports of any GLP Study or clinical trial of such Other Product conducted by or on behalf of Cidara or Cidara-Controlled Affiliates that are received by Cidara or Cidara-Controlled Affiliates during the applicable Other Product Option Period, and (iii) copies of written advice received by Cidara or Cidara-Controlled Affiliates from, and copies of Product Filings (including INDs) with, Regulatory Authorities regarding the development and Regulatory Approval of such Other Product. No later than […***…] before[…***…] (in either case, an “Other Product Efficacy Trial” of the applicable Other Product), Cidara shall provide Mundipharma with written notice thereof and a summary of […***…] (each, an “Other Product Efficacy Trial Notice”).
(c) Other Product Option Exercise; Other Product Amendment. Subject to the terms and conditions of this Agreement, on an Other Product-by-Other Product basis, Mundipharma may exercise the Other Product Option for an Other Product at any time (but only
one time) during the applicable Other Product Option Period by delivering written notice of such exercise (“Other Product Exercise Notice”) to Cidara; provided that if, and only if, the parties agree to develop such Other Product for […***…], the expansion of the License to include the applicable Other Product […***…] shall become effective only upon payment in full of an Other Product option payment determined in accordance with Section 4.4(c)(ii), mutatis mutandis (“Other Product Option Payment”). Promptly following delivery of the Other Product Exercise Notice for an Other Product, the parties shall negotiate in good faith a written amendment to this Agreement for such Other Product (an “Other Product Amendment”) to which an initial Other Global Development Plan and an initial Other Mundipharma Territory Plan for such Other Product would be attached as exhibits, and which would include, without limitation, the provisions described in Sections 4.4(c)(i) through 4.4(c)(viii) and the final paragraph of Section 4.4(c) (in each case, mutatis mutandis); provided, however, that […***…]. If the parties agree to develop such Other Product for any such New Indication (a “New Other Product Indication”), […***…].
(d) Failure to Exercise Other Product Option or to Execute Other Product Amendment. Section 4.4(d) shall apply, mutatis mutandis, to each Other Product.
(e) Cidara Launch of Other Product in Mundipharma Territory. For clarity, Section 4.4(e), mutatis mutandis, shall apply to any Other Product.
4.6 Right of First Negotiation for Anti-Fungal Products. During the period beginning on the Effective Date and expiring on the seventh (7th) anniversary of First Commercial Sale of Licensed Product in the Mundipharma Territory (the “Anti-Fungal ROFN Term”), if either party desires to seek a Third Party Licensee (defined below) of the right to commercialize any Anti-Fungal Product Controlled by such party or in the case of Mundipharma, its Affiliates, or in the case of Cidara, Cidara-Controlled Affiliates (the “Licensing Party”) in the other party’s Territory, then, subject to Section 13.7(a)(iii) hereof, the Licensing Party shall provide the other party with written notice thereof and hereby grants the other party the exclusive right of first negotiation, exercisable only one time per Anti-Fungal Product and solely during the applicable Anti-Fungal Product ROFN Exercise Period, to obtain an exclusive license with respect to such Anti-Fungal Product in the other party’s Territory (each, an “Anti-Fungal Product ROFN”) in accordance with this Section 4.6. For purposes of this Section 4.6, “Third Party Licensee” means a Third Party to which a party proposes to grant a license to commercialize the applicable Anti-Fungal Product that includes the right to book sales of such Anti-Fungal Product. With respect to each such Anti-Fungal Product for which a party desires to seek a Third Party Licensee in the other party’s Territory during the Anti-Fungal ROFN Term, the other party shall, within […***…] after
receipt of notice thereof from the Licensing Party (the “Anti-Fungal Product ROFN Exercise Period”), notify the Licensing Party in writing either that (a) the other party desires to exercise its Anti-Fungal Product ROFN for such Anti-Fungal Product or (b) the other party does not wish to exercise and therefore rejects such Anti-Fungal Product ROFN. If the other party notifies the Licensing Party within such Anti-Fungal Product ROFN Exercise Period that the other party desires to exercise such Anti-Fungal Product ROFN, the parties shall negotiate exclusively and in good faith for up to […***…] from such notification regarding the terms pursuant to which the Licensing Party would grant the other party such license in the other party’s Territory. Failure by the other party to give notice of its interest or lack of interest in exercising an Anti-Fungal Product ROFN within the applicable Anti-Fungal Product ROFN Exercise Period shall be deemed to constitute a waiver by the other party of such Anti-Fungal Product ROFN. During the applicable Anti-Fungal Product ROFN Exercise Period with respect to an Anti-Fungal Product and, if the other party exercises its Anti-Fungal Product ROFN with respect to such Anti-Fungal Product within such Anti-Fungal Product ROFN Exercise Period, during the applicable exclusive […***…] negotiation period, the Licensing Party shall not offer to, or grant, any Third Party any option, license or other right to commercialize such Anti-Fungal Product in the other party’s Territory. If the other party waives or is deemed to have waived its Anti-Fungal Product ROFN with respect to an Anti-Fungal Product, or if the parties fail to enter into a definitive license agreement with respect to such Anti-Fungal Product in the other party’s Territory within such […***…] negotiation period, then the Licensing Party shall be free to license such Anti-Fungal Product to one or more Third Parties in the other party’s Territory and shall have no further obligation to the other party with respect to such Anti-Fungal Product. Effective as of the expiration of the Anti-Fungal ROFN Term, this Section 4.6 shall terminate and be of no further force or effect.
4.7 Development Diligence. Mundipharma shall use Commercially Reasonable Efforts to conduct Mundipharma Territory-Specific development activities with respect to Lead Indication Product in the Lead Indications in the Mundipharma Territory as are necessary to obtain and maintain Regulatory Approval for Lead Indication Product in the Lead Indications in each of the Major Markets, including the development of the Pediatric Licensing Product; provided that, for clarity, Mundipharma shall not be under any obligation to conduct any development activities that are solely for obtaining or maintaining Regulatory Approval in China for the Lead Indication Product or the Pediatric Licensed Product in the Prophylaxis Indication.
4.8 Regulatory Activities in the Mundipharma Territory.
(a) Product Filings. Mundipharma shall be solely responsible for preparing, filing, obtaining and maintaining all Product Filings for Licensed Products (including Pediatric Licensed Product) in the Mundipharma Territory, and subject to Section 4.10, at Mundipharma’s sole expense; provided, however, that (i) Cidara shall be solely responsible for preparing, filing, obtaining and maintaining all INDs for the Lead Indication Trials in the Mundipharma Territory at Cidara’s sole expense, (ii) Cidara shall be the sole holder of such INDs, and (iii) Cidara shall provide to Mundipharma (A) a copy of each such IND, (B) copies of all draft material communications to the applicable Regulatory Authorities in connection with such INDs for review and comment by Mundipharma reasonably in advance of submission to such Regulatory Authorities, and (C) copies of all communications received by Cidara or its Affiliates from such Regulatory Authorities in connection therewith.
(b) Subject to Section 4.8(a), Mundipharma shall be the sole holder of all Product Filings for Licensed Products in the Mundipharma Territory, provided that Mundipharma shall provide to Cidara for review and comment reasonably in advance of submission to such Regulatory Authorities copies of drafts of INDs and those portions of Modules 1 and 2 of XXXx (together with English translations, if required) proposed to be filed with the applicable Regulatory Authorities in the Major Markets which differ from the latest draft of the NDA or the NDA as filed by Cidara, as applicable, for the same Licensed Product in the US. Mundipharma shall provide to Cidara (A) copies of INDs and of Modules 1 and 2 of XXXx as filed with Regulatory Authorities in the Major Markets, and (B) copies of all draft material communications to the applicable Regulatory Authorities in the Major Markets in connection with such INDs and Modules 1 and 2 of XXXx for review and comment by Cidara reasonably in advance of submission of such communications to such Regulatory Authorities. Mundipharma shall promptly provide Cidara with copies, and English summary descriptions, of all material documents, information and correspondence received from any Regulatory Authority in the Major Markets in the Mundipharma Territory relating to Licensed Product and, at Cidara’s reasonable request and expense, copies of any other documents, reports and communications from or to any such Regulatory Authority relating to Licensed Product in the Major Markets.
(c) Regulatory Dossier Development Plan. Within six (6) months of the Effective Date, the parties shall negotiate in good faith to agree a regulatory registration dossier development plan comprising the parties’ plans and timelines for the authoring, co-ordination and submission of XXXx to the FDA, EMA and NMPA, and the coordination of communications with a Regulatory Authority in the Mundipharma Territory with which both Mundipharma has commenced pre-MAA submission communications and Cidara is communicating in relation to an IND for a Lead Indication Trial (the “Regulatory Dossier Development Plan”).
(d) Orphan Drug Designation. Mundipharma shall be solely responsible for preparing, filing, obtaining and maintaining any orphan drug designation for Licensed Product in the Mundipharma Territory, including deciding whether or not such filing(s) will be submitted in any particular country or regulatory jurisdiction. Mundipharma shall provide Cidara with copies of any such orphan drug designation filings. Cidara shall provide to Mundipharma reasonable cooperation and informal, non-financial assistance in connection with any such orphan drug designation, including (in editable format wherever possible): (i) all documentation and other materials submitted by Cidara to the EMA prior to the Effective Date regarding orphan drug designation in respect of the Licensed Product; and (ii) any Data generated by or on behalf of Cidara subsequent to the submission of such documentation and materials to the EMA that is necessary or useful for preparing, filing, obtaining or maintaining any orphan drug designation in the Mundipharma Territory (to the extent not previously provided to Mundipharma).
(e) Costs and Expenses of Regulatory Activities. Except as expressly set forth in this Section 4.8 and in Section 4.10, Mundipharma shall bear all costs and expenses incurred in connection with regulatory activities with respect to Licensed Product in the Mundipharma Territory, and Mundipharma shall have responsibility, and shall be the primary contact, for all interactions with Regulatory Authorities in the Mundipharma Territory with respect to Licensed Product and for all compliance filings, certificates, and, subject to Section 4.15, safety reporting, with respect to Licensed Product in the Mundipharma Territory.
4.9 Regulatory Diligence. Subject to Successful Completion of the applicable Lead Indication Trial, Mundipharma shall use Commercially Reasonable Efforts to obtain and maintain Regulatory Approvals for Lead Indication Product for the corresponding Lead Indication in each of the Major Markets; provided that, for clarity, Mundipharma shall not be under any obligation to obtain or maintain any Regulatory Approvals for the Lead Indication Product or the Pediatric Licensed Product in the Prophylaxis Indication in China.
4.10 CMC Development Activities.
(a) CMC Development Plan. CMC development activities, other than those contained in the Global Development Plan, to be undertaken by or on behalf of Cidara (which may include activities in the Mundipharma Territory that will be conducted by Mundipharma and/or its Affiliates) and which are necessary or useful during the development and/or commercial life of the Licensed Product, including CMC activities intended to improve the manufacturing process(es) for Compound and/or Licensed Product and to reduce the Cost of Goods, shall be contained in a written plan, the initial version of which is attached hereto as Exhibit D (the “CMC Development Plan”). In the event the CMC Development Plan conflicts with the CMC terms set forth in the Global Development Plan, the terms in the Global Development Plan shall prevail.
(b) Conduct of the CMC Development Plan. Cidara shall use Commercially Reasonable Efforts to conduct, or have conducted, the CMC Development Plan in accordance with the budgeted costs and timelines. Upon Cidara’s reasonable request, Mundipharma shall provide reasonable cooperation and informal, non-financial (other than as specified in Section 4.10(e) and Section 5.3 below) assistance to Cidara in connection with Cidara’s management and execution of the CMC Development Plan. Each party shall keep the JMC regularly informed of the status, progress and results of all CMC Development Plan activities conducted by it or on its behalf, and shall discuss in good faith, and attempt to reach consensus regarding, any proposed modification to the CMC Development Plan.
(c) CMC Cost of Goods Reductions. Cidara and Mundipharma shall use Commercially Reasonable Efforts to collaborate to reduce the Cost of Goods to USD $[…***…] per naked vial of Lead Indication Product by […***…].
(d) Change Control. Within six (6) months of the Effective Date, the parties shall negotiate and agree a written change control procedure for the management, cost, discussion and agreement (if required) of any amendment, variation or addition to the CMC-related portions of Regulatory Approvals for the Licensed Product in the Field in the Mundipharma Territory that is proposed by either party or that is requested by any Regulatory Authority in the Mundipharma Territory; provided that after the parties have executed a quality agreement in accordance with the principles as set forth in the Key Supply Terms attached hereto as Exhibit G, any change governed by such quality agreement shall be determined in accordance with the provisions thereof.
(e) Costs and Expenses. Cidara shall:
(i) be solely responsible for all costs and expenses related to the conduct of the CMC Development Plan (excluding those CMC activities included in the Global Development Plan, which shall be subject to Section 6.1(a), and as otherwise subject to the terms relating to costs and expenses agreed as part of a change control procedure agreed pursuant to Section 4.10(d)), provided that Mundipharma shall contribute to the costs of the conduct of the CMC Development Plan by payment of Development Milestone Payment 1 pursuant to Section 5.3; and
(ii) provide (and use Commercially Reasonable Efforts to procure that its CMOs for Licensed Product provide) Mundipharma, at Mundipharma’s reasonable request, with reasonable cooperation and non-financial assistance in connection with CMC regulatory matters arising in the Mundipharma Territory prior to and following the first Regulatory Approval in the Mundipharma Territory, which assistance may include facilitating direct communication between Mundipharma and Third Party contractors of Cidara. After the first Regulatory Approval of Licensed Product in the Mundipharma Territory, Cidara shall provide up to […***…] FTE hours of assistance by Cidara personnel per year in connection with such CMC regulatory matters free of charge to Mundipharma. In the event that Mundipharma requests assistance after the first Regulatory Approval of Licensed Product in the Mundipharma Territory exceeding […***…] FTE hours per year, Mundipharma shall compensate Cidara in respect of such excess time spent in accordance with a reasonable hourly rate to be mutually agreed by the parties. If at any time prior to the first Regulatory Approval for Licensed Product in the Mundipharma Territory, Cidara becomes concerned that the level of assistance being requested by Mundipharma from Cidara personnel is excessively burdensome on Cidara, upon the request of Cidara, the matter shall be referred to the JDC (prior to the first Regulatory Approval of Licensed Product in the Mundipharma Territory) or JMC (after the first Regulatory Approval of Licensed Product in the Mundipharma Territory), and the applicable Subcommittee shall discuss such concerns and attempt in good faith to arrive at a reasonable solution. For any such assistance requested by Mundipharma from Cidara’s CMOs, whether prior to or after the first Regulatory Approval of Licensed Product in the Mundipharma Territory, Mundipharma shall reimburse Cidara for the actual amount invoiced by such CMOs to Cidara for such assistance.
(f) Notwithstanding, the foregoing, the parties may mutually agree that certain activities in the CMC Development Plan be conducted by or on behalf of Mundipharma. In such event, the parties shall update the CMC Development Plan and Mundipharma shall be responsible for such obligations to the same extent as Cidara is obligated hereunder, mutatis mutandis.
4.11 Access to Regulatory Filings.
(a) Mundipharma shall promptly provide to Cidara true and complete copies (including English translations if required) of all ReCoRDs, summaries of product characteristics (SmPCs) and/or national prescribing information for Licensed Product (including Pediatric Licensed Product) as approved by each Regulatory Authority in the Major Markets, and copies of all INDs, master XXXx and Regulatory Approvals for Licensed Product (including Pediatric Licensed Product) in the Mundipharma Territory. Mundipharma hereby grants to Cidara Rights of Reference to all such Product Filings (excluding Expanded Licensed Product Clinical Efficacy Data generated by or on behalf of Mundipharma as an Independent Development Party unless the parties have executed an Expanded Licensed Product Amendment with respect to the applicable Expanded Licensed Product) for the purposes of: (i) obtaining and maintaining Regulatory Approvals for Licensed Product (including Pediatric Licensed Product) in the Cidara Territory, subject to Section 2.3; (ii) to the extent Mundipharma or any of its Affiliates is manufacturing, or
having its CMO manufacture, Compound or Licensed Product, conducting or having conducted CMC activities in relation to Compound or Licensed Product (including Pediatric Licensed Product); (iii) to the extent Mundipharma or any of its Affiliates is manufacturing, or having its CMO manufacture, Compound or Licensed Product, the manufacture of Compound or Licensed Product (including Pediatric Licensed Product) for use or distribution anywhere in the world, subject to Section 2.7; and (iv) complying with applicable pharmacovigilance and other regulatory requirements with respect to Product in the Cidara Territory. For clarity, Cidara shall not have Rights of Reference to such Product Filings for Licensed Product for the purposes of obtaining or maintaining Regulatory Approvals for Product that is not Licensed Product, except that Cidara shall have such Rights of Reference to Data regarding Pediatric Licensed Product that is contained in such Product Filings for the purposes of obtaining or maintaining Regulatory Approvals for Product that is not Licensed Product.
(b) Cidara shall promptly provide to Mundipharma true and complete copies of all Product Filings for Licensed Product (including Pediatric Licensed Product) in the Cidara Territory. Subject to Section 5.3, Cidara hereby grants to Mundipharma the Rights of Reference to all such Product Filings (excluding Expanded Licensed Product Clinical Efficacy Data generated by or on behalf of Cidara unless the parties have executed an Expanded Licensed Product Amendment with respect to the applicable Expanded Licensed Product) for the purposes of: (i) obtaining and maintaining Regulatory Approvals for Licensed Product (including Pediatric Licensed Product) in the Mundipharma Territory, subject to Section 2.5; (ii) conducting or having conducted CMC activities in relation to Licensed Product (including Pediatric Licensed Product); and (iii) complying with applicable pharmacovigilance and other regulatory requirements with respect to Licensed Product (including Pediatric Licensed Product) in the Mundipharma Territory.
(c) Each party shall, promptly upon request of the other party, file with applicable Regulatory Authorities such letters of authorization, access or cross-reference as may be necessary to accomplish the intent of this Section 4.11, which cooperation shall include the provision to Mundipharma by Cidara of the necessary certificates of pharmaceutical product, ancillary documents and supporting information (e.g., on reference pricing) required or requested by a Regulatory Authority in the Mundipharma Territory.
4.12 Regulatory Cooperation.
(a) Each party shall:
(i) use Commercially Reasonable Efforts to provide the other party with all reasonable cooperation and informal, non-financial assistance and take all actions reasonably requested by such other party, without changing the allocation of responsibilities set forth in this Article 4, that are necessary or desirable to enable: (i) Mundipharma to obtain and maintain Regulatory Approvals in the Mundipharma Territory for Licensed Product (including the Pediatric Licensed Product) for the Lead Indications and Expanded Licensed Products that are the subject of Expanded Licensed Product Amendments; and (ii) Cidara to obtain and maintain Regulatory Approvals in the Cidara Territory for Licensed Product (including the Pediatric Licensed Product) for the Lead Indications and Expanded Licensed Products that are the subject of Expanded Licensed Product Amendments;
(ii) to cooperate with any inspection by the FDA, EMA, NMPA or other Regulatory Authority relating to Licensed Product (including Pediatric Licensed Product), including, but not limited to, any inspection prior to approval of an application for Regulatory Approval for Licensed Product (including Pediatric Licensed Product); and
(iii) in a timely manner keep the JSC and JDC reasonably and regularly informed of the status, progress and results of regulatory activities in relation to Licensed Product (including the Pediatric Licensed Product) in the Field in its respective Territory.
(b) Amendments to Regulatory Approvals. Within six (6) months of the Effective Date, the parties shall negotiate and agree on procedures for the management, discussion and agreement (if required) of any amendment, variation or addition to Product Filings and Regulatory Approvals for the Licensed Product in the Field in the Mundipharma Territory (excluding the CMC-related portions thereof, changes to which shall be governed by Section 4.10(d)) that is proposed by either party or that is requested by any Regulatory Authority in the Mundipharma Territory, including without limitation any labelling- or safety-related changes.
4.13 Compliance. In conducting any activity pursuant to this Article 4, each party shall comply with all Applicable Laws, including, as applicable, GLP, GCP, GMP and GVP.
4.14 Records. In conformity with standard pharmaceutical and biotechnology industry practices and the terms and conditions of this Agreement, each party shall prepare and maintain, or shall cause to be prepared and maintained, complete and accurate written records, accounts, notes, reports and data (including Data) with respect to all development and CMC activities of Licensed Product in the Field. Such records shall fully and properly reflect all work done and results achieved in the performance of the development and CMC activities in good scientific manner appropriate for regulatory and patent purposes. Each party shall document all GLP Studies and clinical trials in formal written study records according to Applicable Law, including applicable national and international guidelines such as ICH, GMP, GCP, GLP and GVP. Each Party may review such records (including, to the extent permitted by applicable data privacy laws, clinical study reports and case report forms) maintained by the other party at reasonable times, and upon reasonable notice, to obtain access to the original records to the extent such party has a license to use the Information, Data or Inventions contained in such records pursuant to the terms of this Agreement.
4.15 Global Safety Database; Pharmacovigilance; Adverse Event Reporting.
(a) Pharmacovigilance Procedures. Prior to the execution of a pharmacovigilance agreement regarding Licensed Product (“Pharmacovigilance Agreement”), the parties shall coordinate their pharmacovigilance procedures in connection with the development of the Licensed Product, and Cidara shall submit to Regulatory Authorities in the Mundipharma Territory all safety information and reporting in relation to the Lead Indication Trials in a manner that meets reporting requirements under Applicable Laws in the Mundipharma Territory. Mundipharma shall not conduct any development activities, or engage any Third Party contractor to perform any development activities on its behalf, prior to the execution of the Pharmacovigilance Agreement.
(b) Pharmacovigilance Agreement. Within six (6) months of the Effective Date and in any event prior to initiation of any development activities by Mundipharma, the parties shall negotiate in good faith and enter into a Pharmacovigilance Agreement, which shall include mutually acceptable procedures governing the collection, investigation, reporting, and exchange of information concerning adverse drug reactions/experiences, pregnancy reports and any other information concerning the safety of the Licensed Product, sufficient to permit each party to comply with its regulatory and other legal obligations within the applicable timeframes. The terms and conditions of the Pharmacovigilance Agreement shall be in accordance with U.S., EU and ICH guidelines, except where said guidelines may conflict with existing local regulatory or safety reporting requirements, in which case local reporting requirements shall prevail.
(c) Responsibility. The Pharmacovigilance Agreement, shall specify that, during the Term, each party shall, in its respective Territory, be responsible for monitoring all clinical experiences with respect to Licensed Product in the course of its own Licensed Product development activities. The parties respective responsibilities regarding the following shall also be set forth in the Pharmacovigilance Agreement: (i) filing all required reports with respect thereto (including quality complaints, adverse events and safety data); and (ii) responding to safety issues and to all associated requests of Regulatory Authorities relating to Licensed Product; provided that Cidara shall be responsible for all monitoring and reporting obligations in respect of the Lead Indication Trials, including any reporting required to the appropriate Regulatory Authorities in the Mundipharma Territory.
(d) Global Safety Database, Signaling, Aggregate Reporting and Labelling. As between the parties, Cidara will hold, solely own and be solely responsible for maintaining the global safety database for Licensed Product, provisions concerning which shall be included in the Pharmacovigilance Agreement. In addition, Cidara shall be responsible for the preparation and maintenance of the global developmental safety update reports, periodic safety update reports, risk management plans, signal detection activities, and the company core data sheet in respect of the Licensed Product. Pursuant to the terms of the Pharmacovigilance Agreement, Mundipharma will collaborate with Cidara in respect of signaling, aggregate reporting and labelling activities.
(e) Qualified Person for Pharmacovigilance and Pharmacovigilance System Master File. Subject to the terms of the Pharmacovigilance Agreement, Mundipharma shall have a European Economic Area Qualified Person for Pharmacovigilance, and shall develop and maintain the pharmacovigilance system master file. Cidara shall provide reasonable cooperation and informal, non-financial assistance in connection with the same, including the provision of all relevant pharmacovigilance information for inclusion in the pharmacovigilance system master file. In particular, Cidara shall provide to Mundipharma at least on a quarterly basis a summary report of serious adverse events, aggregate reports (e.g., DSURs, PSURs, RMPs, all as defined by the EMA), investigator brochures and any updates to the same.
(f) Adverse Event and other Regulatory Reporting. As between the parties, following execution of the Pharmacovigilance Agreement: (i) Mundipharma shall be responsible, at its own cost and expense, for the timely reporting of all adverse drug reactions/experiences, product quality complaints and safety data relating to Licensed Product to the appropriate Regulatory Authorities in the Mundipharma Territory (except with respect to any of the foregoing arising in the Lead Indication Trials, which Cidara shall be responsible for reporting to the appropriate Regulatory Authorities in the Mundipharma Territory); and (ii) Cidara shall be responsible, at its own cost and expense, for reporting all adverse drug reactions/experiences, product quality complaints and product safety data relating to Licensed Product to the appropriate Regulatory Authorities in the Cidara Territory; in each case ((i) and (ii)) in accordance with Applicable Laws of the relevant countries and Regulatory Authorities. Each party shall use Commercially Reasonable Efforts to ensure that its Affiliates, licensees and Sublicensees comply with such reporting obligations.
(g) Audits and Inspections. Subject to the terms of the Pharmacovigilance Agreement: (i) either party may audit relevant elements of the other party’s pharmacovigilance system to verify compliance with such party’s pharmacovigilance obligations; and (ii) if either party is subject to inspection of their pharmacovigilance system by a Regulatory Authority, the parties will collaborate in preparing for inspection and addressing any inspection questions and findings, detail of such collaboration to be contained in the Pharmacovigilance Agreement.
4.16 Commercialization in the Mundipharma Territory.
(a) Subject to the terms and conditions of this Agreement, Mundipharma shall control and be solely responsible, at its expense, for marketing, promotion and other commercialization of Licensed Product in the Field in the Mundipharma Territory, including obtaining and maintaining pricing and reimbursement Regulatory Approvals, conducting health economic outcomes research and market research, determining launch sequencing and, subject to Mundipharma’s compliance with its diligence obligations under this Agreement, deciding whether or not to launch in a particular market.
(b) Subject to obtaining Regulatory Approval for the Lead Indication Product in a Lead Indication in a Major Market, Mundipharma shall use Commercially Reasonable Efforts to commercialise the Lead Indication Product for the applicable Lead Indication in such Major Market.
4.17 Named Patient Supply. Prior to the first NDA approval for Licensed Product in the U.S., neither party shall support any named patient program for Licensed Product in such party’s Territory without prior JSC approval, and any such proposed named patient program of a party shall always require unanimous approval of the JSC without resort to the dispute resolution provisions set forth in Section 3.1(f). After the first NDA approval for Licensed Product in the U.S., each party may support named patient programs for Licensed Product in such party’s Territory subject only to discussion by the JSC.
4.18 Manufacturing and Supply.
(a) General. Subject to the terms and conditions of this Agreement and any Commercial Supply Agreement, Cidara shall sell and supply, or cause to be supplied, to Mundipharma, and Mundipharma shall purchase from Cidara: (i) Mundipharma’s, its Affiliates’ and its and their Sublicensees requirements of Licensed Product for clinical trials and other development and registration activities in the Mundipharma Territory, as described in additional detail in Sections 4.18(b) and 4.18(d); and (ii) Mundipharma’s, its Affiliates’ and its and their Sublicensees requirements of Licensed Product for commercial distribution in the Mundipharma
Territory, as described in additional detail in Sections 0 and 4.18(d). Mundipharma acknowledges that […***…], and that accordingly, except as set forth in Section 4.18(b) and in the Key Supply Terms attached hereto as Exhibit G (in the case of Licensed Product clinical trial material), […***…]. Initially, and until otherwise mutually agreed by the parties in writing, […***…]. The parties may subsequently mutually agree in writing that […***…].
(b) Clinical Supply. Cidara shall manufacture, or have manufactured, and supply, or have supplied, to Mundipharma, Licensed Product in final packaged and labeled form for use in clinical trials and other development and registration activities with respect to Licensed Product in the Mundipharma Territory, in accordance with a clinical supply plan and associated terms governing such supply and purchase, including in relation to (i) shelf life, (ii) lead time, (iii) inspection/acceptance and rejection/defects and latent defects, (iv) warranties, (v) exclusions and limitations of liability, and (vi) indemnities; such plan to be developed and unanimously approved by the JSC, without resort to either party’s final decision-making authority, and to be consistent with, and designed to permit Cidara to comply with its obligations under, Cidara’s corresponding supply agreements with its CMOs (the “Clinical Supply Plan”). In addition, the Clinical Supply Plan shall […***…], and Cidara shall […***…], provided that […***…]; provided, however, that the foregoing provisions of this sentence shall not apply to the extent […***…]. The parties agree that clinical supplies of Licensed Product shall be delivered DAP Incoterms 2010 (named European destination), and Cidara shall be responsible for obtaining any approvals required for the import and/or export of clinical supplies in the Mundipharma Territory.
(c) Commercial Supply. Commencing promptly after the Effective Date, the parties, primarily via the JMC, shall work together in good faith to negotiate commercial supply agreements (which shall include providing Mundipharma with reasonable advance notice of all scheduled meetings and scheduled calls with Cidara CMOs so that Mundipharma has an
opportunity to physically attend such meetings and join such calls, and permitting Mundipharma to review and jointly prepare all drafts of term sheets and agreements, and to conduct quality audits and technical assessments) with Cidara’s selected Third Party CMOs (“Cidara CMO(s)”) for Compound (including Compound starting material) and Licensed Product for clinical use and commercial distribution in the parties’ respective Territories (the “CMO Supply Agreements”). As soon as is reasonably practicable following the Effective Date, the parties shall negotiate in good faith and enter into a separate written commercial supply agreement, pursuant to which, subject to Sections 0 and 4.18(d), Cidara will manufacture, or have manufactured, and supply, or have supplied, to Mundipharma, Licensed Product (including Pediatric Licensed Product) for commercial distribution in the Mundipharma Territory (the “Commercial Supply Agreement”). The Commercial Supply Agreement shall be negotiated in good faith by the parties and shall be on commercially reasonable terms consistent with the terms of this Agreement. In any event, but save as provided in the paragraph below, the provisions of the Commercial Supply Agreement shall be consistent with, and shall be designed to permit Cidara to comply with its obligations under, the corresponding CMO Supply Agreements. Subject to the foregoing, the Commercial Supply Agreement, and, to the extent applicable, the CMO Supply Agreements, shall be negotiated in line with the principles set forth in the Key Supply Terms attached hereto as Exhibit G.
In addition, the Commercial Supply Agreement shall […***…], and Cidara shall […***…], provided that […***…]; provided, however, that the foregoing provisions of this sentence shall not apply to the extent […***…].
(d) Supply Price. The transfer price for Licensed Product supplied by or on behalf of Cidara pursuant to this Agreement, the Clinical Supply Plan or the Commercial Supply Agreement (i) for use in clinical trials and other development and registration activities, including any Excluded Validation Batch, shall be equal to […***…] or (ii) for commercial distribution shall be equal to […***…] (in each case, the “Supply Price”).
5. | PAYMENTS |
5.1 Upfront License Payment. Within 10 Business Days of the Effective Date, Mundipharma shall pay to Cidara a non-refundable, non-creditable upfront payment in the amount of USD $30 million (the “Upfront Payment”).
5.2 Global Development Costs. Subject to Section 6.1(a), Cidara and Mundipharma shall share equally (50/50) all Global Development Costs until such time as (a) Mundipharma has
paid or reimbursed an aggregate of $31.207 million of Global Development Costs or (b) all GLP Studies, clinical trials and CMC development activities in the Global Development Plan have been completed or terminated, whichever occurs sooner, and after which time Cidara shall be solely responsible for 100% of Global Development Costs.
5.3 Development Milestone Payments. Within 45 days following the first achievement of each of the milestone events set forth in the table below (each, a “Development Milestone Event”), Cidara shall provide Mundipharma with written notice of such achievement, and Mundipharma shall pay to Cidara the corresponding milestone payment set forth in such table (each, a “Development Milestone Payment”):
Development Milestone Event | Payment (USD) |
1. […***…] | $11.145 million |
2. Successful Completion of ReSTORE Trial | $[…***…] |
3. Successful Completion of ReSPECT Trial | $[…***…] |
Cidara shall deliver top-line results of each Lead Indication Trial to Mundipharma within 30 days after the first availability of such top-line results, and within 15 days after such delivery, the JSC shall convene a meeting to discuss such results. Cidara shall deliver a copy of the final report of each Lead Indication Trial within 30 days after the first availability of such report.
For clarity, each Development Milestone Payment shall be payable only once, for the first achievement of the applicable Development Milestone Event. Development Milestone Payment 1 shall be: (a) fully creditable toward the future royalties payable by Mundipharma hereunder until fully credited, provided that such royalties shall not be reduced in any calendar quarter as a result of such credits by more than […***…]%; and (b) refundable, and refunded (to the extent not already credited toward royalties payable hereunder) on the earlier of (i) December 31, 2024 and (ii) the earlier termination of this Agreement by Mundipharma for Cidara’s uncured material breach pursuant to Section 10.2(a)(i) or by Mundipharma for any reason pursuant to Section 10.3. Development Milestone Payments 2 and 3 are non‑creditable and non‑refundable; provided, however, that:
(1) if, after Mundipharma’s payment of Milestone Payment 2 to Cidara, the final report of the ReSTORE Clinical Trial contains results in the assessment of Global Cure at […***…] of subjects who are randomized to Licensed Product compared to subjects who are randomized to intravenous caspofungin, then […***…]; and
(2) if, after Mundipharma’s payment of Milestone Payment 3 to Cidara, the final report of the ReSPECT Clinical Trial contains results in the assessment of Fungal-Free Survival at […***…] of subjects who are randomized to Licensed Product compared to subjects randomized to the standard antimicrobial regimen, then […***…].
Notwithstanding any other provision of this Agreement to the contrary, including paragraphs (1) and (2) above, if Development Milestone Event 2 or Development Milestone Event 3 is not achieved, and the first approval of the first MAA for Licensed Product for the applicable Lead Indication is received in a Major European Country, then Mundipharma shall pay to Cidara an amount equal to […***…]% of the corresponding Development Milestone Payment upon receipt of such first approval of the first MAA for Licensed Product for such Lead Indication in the first Major European Country (except in the event that […***…]) no later than the applicable due date for the Regulatory/Launch Milestone Payment corresponding to the first approval of the first MAA for Licensed Product for such Lead Indication (in addition to paying to Cidara the applicable Regulatory/Launch Milestone Payment).
5.4 Determination of Milestone Payment Amounts for Major European Country Regulatory Milestone Events. For purposes of Regulatory/Launch Milestone Payments under Section 5.5 that correspond to Acceptance for Filing or approval of an MAA for a Licensed Product in the Major European Countries, the following provisions shall apply. For purposes of this Section 5.4, the EMA shall be deemed to be a “Regulatory Authority of Competent Jurisdiction” in a particular Major European Country if, at the relevant time, either: (i) such Major European Country is an EU member state; or (ii) such Major European Country is not an EU member state but, by treaty, agreement, convention or other understanding with the EU or the EMA, the national government or applicable national Regulatory Authority of such Major European Country recognizes the centralized EU MAA filing procedure, or approval of an MAA filed in accordance therewith (as applicable), to the same extent as if such Major European Country were an EU member state.
(a) If the first MAA for Licensed Product in a particular Major European Country that is Accepted for Filing is (i) filed with the applicable national Regulatory Authority in a particular Major European Country, or (ii) approved by such Regulatory Authority, then the applicable Regulatory/Launch Milestone Event shall be deemed to have been achieved in such Major European Country upon such Acceptance for Filing or receipt of notification of approval by such Regulatory Authority, as applicable, and the amount payable upon such achievement shall be equal to 20% of the corresponding Regulatory/Launch Milestone Payment set forth in the table in Section 5.5.
(b) If the first MAA for Licensed Product in a particular Major European Country that is Accepted for Filing by a Regulatory Authority of Competent Jurisdiction is (i) filed with the EMA using the centralized EU filing procedure, or (ii) approved by the European Commission, then the applicable Regulatory/Launch Milestone Event shall be deemed to have been achieved in such Major European Country upon such Acceptance for Filing or receipt of notification of approval by the European Commission, as applicable, and the amount payable upon
such achievement shall be equal to 20% of the corresponding Regulatory/Launch Milestone Payment set forth in the table in Section 5.5.
For clarity, if the applicable Regulatory/Launch Milestone Event is achieved simultaneously in multiple Major European Countries by the filing or approval of the same MAA, then the amount payable upon such achievement shall be determined by multiplying the corresponding Regulatory/Launch Milestone Payment set forth in the table in Section 5.5 by the fraction X/5, where X equals the number Major European Countries in which such Regulatory/Launch Milestone Event is simultaneously achieved. If the applicable Regulatory/Launch Milestone Event is achieved simultaneously in all Major European Countries by the same MAA, then the full amount of the corresponding Regulatory/Launch Payment shall be payable upon such achievement.
5.5 Regulatory and Launch Milestone Payments. Within 45 days following the first achievement (whether by Mundipharma, an Affiliate of Mundipharma or a Sublicensee) of each of the milestone events set forth in the table below by the Lead Indication Product or other Licensed Product (each, a “Regulatory/Launch Milestone Event”), Mundipharma shall provide Cidara with written notice of such achievement and shall pay to Cidara the corresponding non‑refundable, non‑creditable milestone payment set forth in such table (each, a “Regulatory/Launch Milestone Payment”), subject to Section 5.4:
Regulatory/Launch Milestone Event | Payment (USD) |
1. […***…] | $[…***…]* |
2. […***…] | $[…***…]* |
3. […***…] | $[…***…]* |
4. […***…] | $[…***…]† |
5. […***…] | $[…***…] |
6. […***…] | $[…***…] |
7. […***…] | $[…***…] |
8. […***…] | $[…***…] per country‡ |
* | Aggregate amount payable for achievement in all Major European Countries. |
† | […***…]. |
‡ | $[…***…] in total if Regulatory/Launch Milestone Event achieved in […***…]. |
The aggregate Regulatory/Launch Milestone Payment for each of Regulatory/Launch Milestone Event No. 1, Regulatory/Launch Milestone Event No. 2 and Regulatory/Launch Milestone Event No. 3 shall be payable only once but may be payable in multiple payments as specified in Section 5.4. The Regulatory/Launch Milestone Payment for each of Regulatory/Launch Milestone Event No. 4, Regulatory/Launch Milestone Event No. 5, Regulatory/Launch Milestone Event No. 6 and Regulatory/Launch Milestone Event No. 7 shall be payable only once, for the first achievement of the applicable Regulatory/Launch Milestone Event. The Regulatory/Launch Milestone Payment for Regulatory/Launch Milestone Event No. 8 shall be payable once per listed country (i.e., up to a maximum of […***…] times), for the first achievement of the applicable Regulatory/Launch Milestone Event in each listed country.
5.6 Commercialization Milestone Payments. Within 45 days following the end of the calendar year in which each of the events set forth below (each, a “Net Sales Milestone Event”) is first achieved, Mundipharma shall pay to Cidara the corresponding one-time, non‑refundable, non‑creditable milestone payment (each, a “Net Sales Milestone Payment”) set forth below:
Commercialization Milestone Event | Payment (USD) |
First calendar year in which aggregate annual Net Sales of all Licensed Products in the Mundipharma Territory equal or exceed $[…***…] | $[…***…] |
First calendar year in which aggregate Net Sales of all Licensed Products in the Mundipharma Territory equal or exceed $[…***…] | $[…***…] |
First calendar year in which aggregate annual Net Sales of all Licensed Products in the Mundipharma Territory equal or exceed $[…***…] | $[…***…] |
First calendar year in which aggregate annual Net Sales of all Licensed Products in the Mundipharma Territory equal or exceed $[…***…] | $[…***…] |
First calendar year in which aggregate annual Net Sales of all Licensed Products in the Mundipharma Territory equal or exceed $[…***…] | $[…***…] |
First time aggregate annual Net Sales of all Licensed Products in the Mundipharma Territory equal or exceed $[…***…] | $[…***…] |
Each Net Sales Milestone Payment shall be payable only once. Each Net Sales Milestone Payment shall be paid within 45 days of the end of the calendar year in which the corresponding Net Sales Milestone Event is achieved. If multiple Net Sales Milestone Events are achieved in any given calendar year, the Net Sales Milestone Payments corresponding to all of such multiple Net Sales Milestone Events shall be paid within 45 days of the end of such calendar year.
5.7 Royalties. Subject to Sections 5.8 to 5.11 below, Mundipharma shall pay to Cidara running royalties on aggregate annual Net Sales of all Licensed Product by Mundipharma, its Affiliates and Sublicensees in the Mundipharma Territory at the following incremental royalty rates:
Aggregate Annual Net Sales of Licensed Product in the Mundipharma Territory | Royalty Rate |
On that portion of aggregate annual Net Sales in a calendar year of all Licensed Products in the Mundipharma Territory less than $[…***…] | […***…]% |
On that portion of aggregate annual Net Sales in a calendar year of all Licensed Products in the Mundipharma Territory greater than $[…***…] but less than $[…***…] | […***…]% |
On that portion of aggregate annual Net Sales in a calendar year of all Licensed Products in the Mundipharma Territory greater than $[…***…] but less than $[…***…] | […***…]% |
On that portion of aggregate annual Net Sales in a calendar year of all Licensed Products in the Mundipharma Territory greater than $[…***…] but less than $[…***…] | […***…]% |
On that portion of aggregate annual Net Sales in a calendar year of all Licensed Products in the Mundipharma Territory greater than $[…***…] | […***…]% |
5.8 Royalty Term. Royalties under Section 5.7 shall be payable on a Licensed Product-by-Licensed Product and country-by-country basis from the First Commercial Sale of a Licensed Product in a country until the latest of: (a) […***…] years from such First Commercial Sale of such Licensed Product in such country; (b) expiration of all Regulatory Exclusivity for such Licensed Product in such country; and (c) expiration of the last-to-expire Valid Claim of the Cidara Patents or Joint Patents that would, in the absence of the License (or, in the case of Joint Patents, in the absence of Mundipharma’s joint ownership interest thereof), be infringed by the
5.9
manufacture, use, sale, offer for sale or import of such Licensed Product in such country (the “Royalty Term”). For clarity: (i) there shall be only one Royalty Term per country for the Lead Indication Product, regardless of the number of indications for which the Lead Indication Product may be approved in such country, and such Royalty Term shall begin on the First Commercial Sale of such Lead Indication Product in such country; and (ii) other than an Expanded Licensed Product described in clause (A) of Section 1.47 (which itself is a new indication of any other Licensed Product, including any other Expanded Licensed Product), there shall be only one Royalty Term per country for each Expanded Licensed Product, regardless of the number of indications for which such Expanded Licensed Product may be approved in such country, and such Royalty Term shall begin on the First Commercial Sale of such Expanded Licensed Product in such country; provided, however, that […***…]; in which case, the Royalty Term for such Expanded Licensed Product in such country shall be […***…]. By way of example only, […***…] (x) shall […***…] for the purposes of this Section 5.8, (y) there shall be […***…], and (z) such […***…] in such country (it being understood that […***…]).
5.10 Consequences of Royalty Term Expiry. On a Licensed Product-by-Licensed Product and country-by-country basis, upon expiration of the Royalty Term for a Licensed Product in a country, Mundipharma’s License with respect to such Licensed Product in such country shall become royalty‑free, fully-paid, irrevocable and perpetual.
5.11 Generic Competition. On a country-by-country basis, if, following the launch of a Generic Product during the Royalty Term for Licensed Products in a country, the […***…] in such country reaches the percentage thresholds specified in Section 5.10(a) to 5.10(e) below, then for the remainder of the Royalty Term for Licensed Products in such country, Mundipharma’s royalty rates then applicable with respect to Net Sales of such Licensed Products in such country (i.e., as set forth in Section 5.7 but as such royalties may have been further reduced pursuant to Section 5.11) shall be reduced as follows.
(a) during any calendar quarter in which […***…] in a country in the Mundipharma Territory is equal to or greater than […***…] ([…***…]%) but less than […***…] percent ([…***…]%), the royalty rates for Licensed Products sold in such country shall be reduced to […***…] percent ([…***…]%) of the royalty rates then applicable;
(b) during any calendar quarter in which […***…] in a country in the Mundipharma Territory is equal to or greater than […***…] percent ([…***…]%) but less than […***…] ([…***…]%), the royalty rates for Licensed Products sold in such country shall be reduced to […***…] ([…***…]%) of the royalty rates then applicable;
(c) during any calendar quarter in which […***…] in a country in the Mundipharma Territory is equal to or greater than […***…] ([…***…]%) but less than […***…] percent ([…***…]%), the royalty rates for Licensed Products sold in such country shall be reduced to […***…] ([…***…]%) of the royalty rates then applicable;
(d) during any calendar quarter in which […***…] in a country in the Mundipharma Territory is equal to or greater than […***…] […***…]%) but less than […***…] percent ([…***…]%), the royalty rates for Licensed Products sold in such country shall be reduced to […***…] ([…***…]%) of the royalty rates then applicable; and
(e) during any calendar quarter in which […***…] in a country in the Mundipharma Territory is equal to or greater than […***…] percent ([…***…]%), the royalty rates for Licensed Products sold in such country shall be reduced to […***…] ([…***…]%) of the royalty rates then applicable.
For the purposes of this Section 5.10, […***…] shall mean […***…].
5.12 Third Party Licenses. In the event that Mundipharma or its Affiliate or Sublicensee (as applicable) is required to obtain one or more licenses under Patents of Third Parties (excluding Sublicensees) that are necessary for the manufacture, use, sale, offer for sale or import of Licensed Product in a country of the Mundipharma Territory (“Third Party Licenses”), […***…]% of the royalties actually paid by Mundipharma or such Affiliate or Sublicensee (as applicable) under such Third Party Licenses with respect to sales of such Licensed Product in such country for a calendar quarter will be creditable against the royalties payable by Mundipharma to Cidara with respect to Net Sales of such Licensed Product in such country for such calendar quarter; provided, however, that in no event will the royalties payable by Mundipharma to Cidara hereunder with respect to Net Sales of such Licensed Product in such country for such calendar quarter be reduced by more than […***…]% as a result of any and all such credits in the aggregate (but any portion of the royalties paid under Third Party Licenses with respect to sales of such Licensed Product in such country that Mundipharma would have been entitled to credit against royalties payable to Cidara in the absence of the foregoing limitation on aggregate credits in any calendar quarter shall be carried over and applied against royalties payable to Cidara in respect of such Licensed Product in any country in subsequent calendar quarters until the full deduction is taken); and provided, further, that Mundipharma will not be entitled to credit any portion of royalties that are paid or payable by Mundipharma or its Affiliate or Sublicensee to any Third Party with respect to sales of a Combination Product in any country by reason of the inclusion in such Combination Product of any Other Active.
5.13 Royalty Floor. In no event shall the effective royalty rate applicable to Net Sales of Licensed Products in a country be reduced by more than an aggregate of […***…] percent ([…***…]%) in any calendar quarter as a result of any and all reductions and credits pursuant to Sections 4.4(e), 5.10 and 5.11 ([…***…]) in the aggregate.
6. | PAYMENT; RECORDS; AUDITS |
6.1 Payment; Reports.
(a) Global Development Costs. Within 30 days after the end of each calendar quarter during the performance of the Global Development Plan, each party shall provide to the other a written statement setting forth the Global Development Costs incurred by such first party and its Affiliates during such calendar quarter, such statement to include a reasonably detailed breakdown of the components of such Global Development Costs and the Global Development Plan activities to which such Global Development Costs are attributable (each such statement, a “Cost Report”). For the avoidance of doubt, Mundipharma may not submit a Cost Report to Cidara for Global Development Costs except to the extent the Global Development Plan contemplates Mundipharma incurring such amounts or such amounts have otherwise been approved by the JSC. Following receipt of Mundipharma’s Cost Report, if any, Cidara will provide Mundipharma with a written invoice for Mundipharma’s 50% share of the Global Development Costs for such calendar quarter; provided, however, that from and after such time as Mundipharma has paid or reimbursed an aggregate of $31.207 million of Global Development Costs, Cidara’s obligation to provide Cost Reports and its right to provide invoices to Mundipharma for Global Development Costs shall cease. Within 45 days after delivery of the Cost Report and associated invoice for a calendar quarter, Mundipharma shall pay the invoiced amount to Cidara (less any portion of such amount that Mundipharma disputes in good faith, of which Mundipharma shall promptly notify Cidara). Each party shall respond promptly to the other party’s questions regarding any Cost Report delivered hereunder or reasonable requests for supporting documentation, including, without limitation, copies of agreements or work orders for Global Development Plan activities performed by Third Parties (from which copies such party may redact confidential or proprietary information that is not necessary for the other party to ascertain Global Development Costs).
(b) Royalties. Royalties under Section 5.7 shall be calculated and reported for each calendar quarter and shall be paid within 45 days of the end of the calendar quarter. Each payment of royalties shall be accompanied or preceded by a report of Net Sales in sufficient detail to permit confirmation of the accuracy of the payment made, including, on a Licensed Product-by-Licensed Product and country-by-country basis, the number of each type of Licensed Product sold, gross sales, Net Sales and itemized deductions from gross sales (by major category as set forth in the definition of Net Sales), details of any Net Sales adjustments for any Combination Product, any applicable reductions or adjustments made pursuant to Section 5.8 and/or Section 5.10, details of any royalty credits taken pursuant to Section 5.3 in respect of Development Milestone Payment 1, and Section 5.11 on a Third Party License-by-Third Party License basis, royalties payable, and the exchange rates used, in each case on a Product-by-Product and country-by-country basis.
6.2 Exchange Rate; Manner and Place of Payment. All payment amounts specified in this Agreement are expressed in U.S. dollars, and all payments by Mundipharma to Cidara under this Agreement shall be paid in U.S. dollars. If any currency conversion shall be required in connection with the calculation of amounts payable hereunder, such conversion shall be calculated at the rate of exchange for such currency used throughout Mundipharma’s accounting system in conformity with Accounting Standards for the calendar quarter for which payment is due. All payments owed under this Agreement shall be made by wire transfer to a bank and account designated in writing by Cidara, unless otherwise specified in writing by Cidara.
6.3 Income Tax Withholding and VAT.
(a) Except as set forth herein, each party will pay all taxes (including related interest and penalties) imposed on its share of income, profits or gains arising directly or indirectly from the efforts of, or the receipt of any payment by, such party pursuant to this Agreement.
(b) If any taxes or similar duties, assessments or charges in the nature of a tax due to governmental or tax authorities (including related interest and penalties) (“Taxes”) are required to be withheld by one party (“payor”) from any payment to the other party (“payee”) under this Agreement, the payor shall (i) deduct such Taxes from the payment to the payee, (ii) timely pay such Taxes to the proper governmental or taxing authority, and (iii) send proof of payment to the payee within 30 days following such payment. Any amount actually withheld and remitted by the payor to such governmental or tax authority pursuant to this Section 6.3(b) shall be treated for all purposes of this Agreement as paid to the payee. Mundipharma acknowledges that based on Applicable Law as of the Effective Date no withholding tax is required to be deducted or withheld from payments made by Mundipharma to Cidara pursuant to this Agreement. Subject to Section 6.3(c) below, if the payor makes a payment without deduction for Tax withholding and an amount of Tax should have been withheld from such payment, then the payor shall be entitled to recover the underwithheld Tax by an additional withholding from any amount payable to the payee under this Agreement only if such failure to withhold is solely the fault of the payee (for example, without limitation, where the payor’s failure to properly withhold tax is due to the payee’s provision of incorrect tax information to the payor.
(c) If the payor is required to make a payment to the payee that is subject to a deduction or withholding of Taxes and the obligation to make such deduction or withholding arises or is increased solely as a result of (i) any failure on the part of the payor to comply with Applicable Laws relating to the withholding of Taxes or (ii) the assignment of this Agreement or transfer of any rights or obligations under this Agreement by the payor (a “Withholding Tax Action”), then the payment by the payor in respect of which such deduction or withholding of Taxes is required to be made shall be increased by such amount as will leave the payee in the same net of Tax position it would have been in had the Withholding Tax Action not occurred (it being understood that Taxes that would have been deducted or withheld in accordance with Section 6.3(b) in the absence of a Withholding Tax Action are the payee’s responsibility). Notwithstanding the foregoing, no additional amounts shall be payable by the payor to the payee under this Section 6.3(c) if the payee determines (acting in good faith) that any Taxes arising due to a Withholding Tax Action will be recovered by the payee or will offset the payee’s otherwise payable Taxes for the Tax year in which the Withholding Tax Action occurs.
(d) Notwithstanding anything contained in Section 6.3(a), 6.3(b) or 6.3(c), this Section 6.3(d) shall apply with respect to any value added, sales, use, goods, services, or other similar tax (“Sales Tax”) applicable to any sums payable by one party to the other party pursuant to this Agreement.
(i) All sums expressed to be payable under this Agreement are exclusive of Sales Tax (if applicable).
(ii) If any Sales Tax is payable on or in respect of any supply by any party under this Agreement, the recipient of the supply shall pay the amount of the Sales Tax to the supplier (to the extent that the supplier is liable to account for such Sales Tax to the appropriate tax or governmental authority) in respect of such supply following the receipt of a valid Sales Tax invoice in the appropriate form issued by the supplier in respect of the supply, such Sales Tax to be payable on the later of the due date of the payment for the supply to which such Sales Tax relates and ten (10) Business Days after the receipt by the recipient of the supply of the applicable Sales Tax invoice, and the supplier shall (where it is liable to account for such Sales Tax) timely remit such Sales Tax to the appropriate tax or governmental authority. Each party may propose to the other in writing changes to the payment and invoicing arrangements in relation to the supplied services, with a view to minimising, so far as lawfully and reasonably practicable, costs suffered on account of Sales Tax by that party. If a party receives such a proposal from the other, it will, acting reasonably, consider the proposal and, to the extent that it agrees to the proposal, will provide its reasonable cooperation in implementing it.
(iii) Where a party is required under this Agreement to pay an amount in respect of, or otherwise reimburse, any cost, charge or expense incurred by another party, the first party shall not be required to pay or reimburse any amount in respect of Sales Tax which is recoverable (whether by way of repayment, credit or set off) by the second party, and the second party shall use all reasonable endeavors to seek to minimize irrecoverable Sales Tax.
(iv) If any Sales Tax originally paid by the recipient of the relevant supply for Sales Tax purposes to the supplier in accordance with the terms of this Agreement is in whole or in part subsequently determined (by the applicable tax authority) not to have been chargeable, and the supplier has obtained a refund of such Sales Tax from the relevant tax or governmental authority, the supplier shall pay an amount equal to any such Sales Tax repaid by the relevant tax or governmental authority to the recipient of the relevant supply within ten (10) Business Days of receipt from the tax or governmental authority (whether receipt is by way of repayment, credit or set off).
(e) The parties agree to cooperate with one another and to use reasonable efforts to (I) reduce or eliminate Tax withholding or similar obligations and (II) assist in the claiming of foreign tax credits, refunds, or deductions, in each case, in respect of the payments made by one party to the other party under this Agreement. Without limiting the generality of the foregoing, each party shall (i) upon written request, provide the other with any tax forms and information in such party’s possession (and which it is legally able to provide) that may be reasonably necessary in order for (A) the payor not to withhold Tax, or to withhold Tax at a reduced rate, under an applicable double tax treaty or (B) the payee to obtain the benefit of any present or future applicable double tax treaty, within a reasonable time prior to the date the applicable payment is due, and (ii) provide the other with reasonable assistance to enable the recovery, as permitted by Law, of withholding Taxes, Sales Tax, or similar obligations resulting from payments made under this Agreement, such recovery to be for the benefit of the party bearing such Tax or other obligation.
6.4 Audits.
(a) Global Development Cost Audit. During the performance of any Global Development Plan activities and for a period of […***…] years thereafter, each party and its Affiliates shall keep complete and accurate records pertaining to Global Development Costs incurred by it in sufficient detail to permit the other party to confirm the calculation of Global Development Costs (including, in the case of Cidara, the applicable Cost of Goods) and the accuracy of the other party’s invoices delivered to such party pursuant to Section 6.1(a). Each party shall have the right to cause an independent, certified public accountant reasonably acceptable to the other party to audit such records (other than tax returns and records related to tax returns) to confirm the calculation of such Global Development Costs and the accuracy of the other party’s invoices for such Global Development Costs for a period covering not more than the preceding […***…] full years. The audited party may require such accountant to execute a reasonable confidentiality agreement with the audited party prior to commencing the audit. Such audits may be conducted during normal business hours upon reasonable prior written notice to the party to be audited, but no more frequently than once per year. No accounting period of a party shall be subject to audit more than one time pursuant to this Section 6.4(a). Prompt adjustments (including remittances of underpayments or overpayments disclosed by such audit) shall be made by the parties to reflect the results of such audit, and no later than 30 days after the date of the accountant’s audit report. The auditing party shall bear the full cost of such audit unless such audit discloses an overstatement by the audited party of the audited party’s Global Development Costs in any calendar year of […***…]% or more, in which case the audited party shall bear the full cost of such audit. Alternatively, if a party provides notice of exercise of its audit right under this Section 6.4(a), then, at the other party’s written request made within 30 days of such exercise, the parties shall jointly retain an independent, certified public accountant reasonably acceptable to both parties to conduct an audit of both parties’ Global Development Costs for the applicable period, in which case the parties shall share equally (50/50) the cost of such joint audit. Prompt adjustments (including remittances of underpayments or overpayments disclosed by such audit) shall be made by the parties to reflect the results of such audit, and no later than 30 days after the date of the jointly retained, independent accountant’s audit report. If such jointly initiated audit discloses that one party (but not the other party) has overstated its Global Development Costs in any calendar year covered by such audit by […***…]% or more, such party shall reimburse the other party for the 50% share of the joint audit costs that was initially borne by the other party. The results of such audit will be final, absent manifest error.
(b) CMC Development Cost Audit. During the performance of any CMC Development Plan activities and for a period of […***…] years thereafter, each party and its Affiliates shall keep complete and accurate records pertaining to its external costs incurred by it (including any Costs of Goods) in sufficient detail to permit the other party to verify such costs incurred as compared to the budgeted amounts in the CMC Development Plan. Each party shall have the right to cause an independent, certified public accountant reasonably acceptable to the other party to audit such records (other than tax returns and tax records) to verify such costs for a period covering
(c)
not more than the preceding […***…] full years. The audited party may require such accountant to execute a reasonable confidentiality agreement with the audited party prior to commencing the audit. Such audits may be conducted during normal business hours upon reasonable prior written notice to the party to be audited, but no more frequently than once per year. No accounting period of a party shall be subject to audit more than one time pursuant to this Section 6.4(b). The auditing party shall bear the full cost of such audit.
(d) Net Sales Audits. Mundipharma shall keep, and shall cause its Affiliates and Sublicensees to keep, complete and accurate records pertaining to the sale or other disposition of Licensed Products in sufficient detail to permit Cidara to confirm the accuracy of all payments due hereunder, for at least […***…] full calendar years following the end of the calendar year to which they pertain. Cidara shall have the right, once annually, to cause an independent, certified public accountant reasonably acceptable to Mundipharma to audit such records to confirm Net Sales, royalties and the timing of achievement of Net Sales Milestone Events, for a period covering not more than the preceding […***…] full calendar years; provided, however, that Mundipharma shall not be required to provide, and neither the independent accountant nor the Cidara shall be entitled to review or examine, the tax returns or tax records of Mundipharma, its Affiliates of Sublicensees. No audited period shall be subject to audit under this Section 6.4(c) more than once. Such audits may be exercised during normal business hours upon reasonable prior written notice to Mundipharma, but no more frequently than once per year. The auditor will execute a reasonable written confidentiality agreement with Mundipharma and will disclose to Cidara only such information as is reasonably necessary to provide Cidara with information regarding any actual or potential discrepancies between amounts reported and actually paid and amounts payable under this Agreement, and between the reported timing and actual timing of achievement of Net Sales Milestone Events. The auditor will send a copy of the report to Mundipharma at the same time it is sent to Cidara. The report sent to both parties will include the methodology and calculations used to determine the results. If such audit reveals that Mundipharma has failed to accurately report information pursuant to Section 6.1(b) or to make any Net Sales Milestone Payment (or portion thereof) when actually due under this Agreement, then Mundipharma, within 30 days after receipt of the final audit report, shall pay to Cidara any underpaid amounts due under this Agreement, together with interest on such underpaid or late amounts calculated in accordance with Section 6.5. Cidara shall bear the full cost of such audit unless such audit discloses an underpayment by Mundipharma of more than […***…]% of the amount due for any calendar year under this Agreement, in which case Mundipharma shall bear the full cost of such audit. If such audit discloses an overpayment by Mundipharma, then Mundipharma will deduct the amount of such overpayment from amounts otherwise owed to Cidara under this Agreement. The results of such audit will be final, absent manifest error.
6.5 Late Payments. In the event that any payment due under this Agreement is not made when due, simple interest shall accrue on the late payment at a rate per annum that is […***…] basis points (i.e., […***…] percentage points) above the then-current prime rate quoted by Citibank in New York City (or such other rate and source as the parties mutually agree in writing) for the period from the due date for payment until the date of actual payment; provided, however, that
(a) in no event shall such rate exceed the maximum legal annual interest rate;
(b) the payment of such interest shall not limit a party from exercising any other rights it may have as a consequence of the lateness of any payment that it is due; and
(c) The parties may agree that a delay to a payment arising solely due to the parties’ efforts to lawfully reduce Tax or Sales Tax applicable to such payment shall not be a late payment that is subject to this Section 6.5, such agreement not to be unreasonably withheld. Any such agreement shall be in writing and delivered in accordance with the terms of this Agreement.
7. | CONFIDENTIALITY |
7.1 Confidential Information. Subject to Section 7.3, “Confidential Information” of a party shall mean any Information or Business Information furnished by or on behalf of such party (the “Disclosing Party”) to the other party (the “Receiving Party”) pursuant to this Agreement or under the Prior CDA, whether in written, oral, visual, electronic or other form. Notwithstanding the foregoing definition, the parties hereby agree that the following information shall be deemed to constitute the “Confidential Information” of each party (without regard to which party initially disclosed such information to the other party), and that each party shall be deemed both a “Receiving Party” and a “Disclosing Party” with respect thereto, for purposes of this Agreement:
(a) the Global Development Plan, the Mundipharma Territory Plan and the CMC Development Plan;
(b) if applicable, in each case, the Expanded Licensed Product Global Development Plan for each Expanded Licensed Product, the Expanded Licensed Product Mundipharma Territory Development Plan for each Expanded Licensed Product, the Subcutaneous Global Development Plan, the Subcutaneous Mundipharma Territory Development Plan, and any plan equivalent to any of the foregoing with respect to an Other Product as contemplated by Section 4.5;
(c) the agenda and minutes of each meeting of the JSC or any Subcommittee (including, in each case, all accompanying documents, exhibits and attachments thereto) and all reports and written disclosures provided by either party to the JSC or any Subcommittee (or to the other party’s representatives on the JSC or any Subcommittee);
(d) any amendment to this Agreement;
(e) Information and Data generated pursuant to any of the plans described in Sections 7.1(a) and 7.1(b);
(f) all written materials provided by either party to […***…] pursuant to Section 3.1(f)(iv); and
(g) all written determinations, recommendations and awards issued by […***…], arbitral tribunal or Independent Expert (including any Expert Forecast) pursuant to this Agreement.
7.2 Obligations of Confidentiality and Non-Use. Except to the extent expressly authorized by this Agreement or otherwise agreed in writing by the parties, the Receiving Party
7.3
agrees that, during the Term and for seven (7) years thereafter, the Receiving Party shall keep confidential and shall not publish or otherwise disclose and shall not use for any purpose, other than as expressly provided for in this Agreement, any Confidential Information of the Disclosing Party. The Receiving Party may use Confidential Information only to the extent required to exercise its rights and fulfil its obligations under this Agreement. The Receiving Party shall use at least the same standard of care as it uses to protect proprietary or confidential information of its own, but no less than reasonable care, to ensure that its, and its Affiliates’, directors, officers, employees, agents, consultants and other representatives (“Representatives”) do not disclose or make any unauthorized use of the Confidential Information. Receiving Party shall be responsible for any failure by its Representatives which, if committed by Receiving Party, would be a breach of this Agreement. The Receiving Party will promptly notify the Disclosing Party upon discovery of any unauthorized use or disclosure of the Confidential Information.
7.4 Exceptions. Confidential Information shall not include any information that the Receiving Party can demonstrate: (a) is, or hereafter becomes, through no act or failure to act on the part of the Receiving Party in breach of this Agreement, generally known or available in the public domain; (b) is known by the Receiving Party at the time of receiving such information, as evidenced by its contemporaneously-maintained written records, with no restrictions on its use or disclosure; (c) is hereafter furnished to the Receiving Party on a non-confidential basis by a Third Party, as a matter of right (i.e., without breaching any obligation such Third Party may have to the Disclosing Party); or (d) is independently discovered or developed by the Receiving Party, independently of the activities undertaken by the Receiving Party pursuant to this Agreement and without the use of, reliance on, or reference to Confidential Information of the Disclosing Party, as evidenced by the Receiving Party’s contemporaneously-maintained written records.
7.5 Authorized Disclosure. Each party may disclose Confidential Information of the other party only as expressly permitted by this Agreement, or if and to the extent such disclosure is:
(a) made by the Receiving Party to a patent authority as may be reasonably necessary for the purposes of filing, prosecuting or enforcing Patents as permitted by this Agreement;
(b) necessary in order to enforce such party’s rights under this Agreement and/or to perform its obligations under this Agreement;
(c) necessary in order to prosecute or defend litigation as permitted by this Agreement;
(d) made in response to a valid order of a court of competent jurisdiction or other competent authority, or otherwise required by applicable laws, rules or regulations, or the listing rules of any exchange on which such party’s securities are traded;
(e) necessary in Product Filings that the Receiving Party has the right to file, or holds, as expressly set forth in this Agreement;
(f) to the Receiving Party’s Affiliates, licensees and sublicensees/Sublicensees, potential licensees and sublicensees/Sublicensees, and to the Receiving Party’s and its Affiliates’ Representatives who, in each case, need to know such Confidential Information in order for the Receiving Party to exercise its rights or fulfill its obligations under this Agreement, provided, in each case, that any such Affiliate, actual or potential licensee or sublicensee/Sublicensee, or Representative is bound by obligations of confidentiality and non-use at least as restrictive as those set forth in this Article 7; and
(g) disclosure to Third Parties in connection with due diligence investigations by such Third Parties, and disclosure to potential Third Party investors in confidential financing documents, provided, in each case, that any such Third Party needs to know such Confidential Information and is bound by reasonable obligations of confidentiality and non-use.
Notwithstanding the foregoing, in the event the Receiving Party is required to make a disclosure of the Disclosing Party’s Confidential Information pursuant to Section 7.4(c) or 7.4(d), it will, to the extent practicable and not prohibited by law, (i) give reasonable advance notice to the Disclosing Party of such disclosure, (ii) provide the Disclosing Party with the opportunity to secure confidential treatment of such Confidential Information to be disclosed; (iii) cooperate with any such efforts by the Disclosing Party, at the Disclosing Party’s request and expense, to secure confidential treatment of such Confidential Information; and (iv) disclose only such minimal portion of the Confidential Information as is, on the advice of counsel, required to be disclosed. Disclosure by the Receiving Party of Confidential Information in accordance with any of the foregoing provisions of this Section 7.4 shall not, in and of itself, cause the information so disclosed to cease to be treated as Confidential Information under this Agreement, except to the extent that, by virtue of disclosure by the Receiving Party in full compliance with this Section 7.4, such information becomes generally known or available.
7.6 Confidentiality of this Agreement. Except as otherwise provided in this Article 7, each party agrees not to disclose to any Third Party the terms of this Agreement without the prior written consent of the other party hereto, except that each party may disclose the terms of this Agreement that are otherwise made public as contemplated by Section 7.6 or to the extent such disclosure is permitted under Section 7.4.
7.7 Public Announcements.
(a) The parties have agreed upon the content of a press release which shall be issued by Cidara (or jointly by the parties, if mutually agreed) substantially in the form attached hereto as Exhibit H, the release of which the parties will coordinate in order to accomplish the same promptly upon execution and delivery of this Agreement. Except to the extent already disclosed in a press release or other public communication issued in accordance with this Agreement, no public announcement concerning this Agreement, its subject matter or the transactions described herein shall be made, either directly or indirectly, by either party or its Affiliates, except as may be required by applicable law (including disclosure requirements of the U.S. Securities and Exchange Commission (“SEC”)), judicial order, or stock exchange or quotation system rule without first obtaining the approval of the other party and agreement upon the nature, text and timing of such announcement, which approval and agreement shall not be unreasonably withheld or delayed. The party desiring to make any such voluntary public announcement shall provide the other party with a written copy (including by email) of the proposed announcement in reasonably sufficient time prior to public release to allow the other party to comment upon such announcement, prior to public release. In the case of press releases or other public communications required to be made by law, judicial order or stock exchange or quotation system rule, the party making such press release or public announcement shall provide to the other party a copy of the proposed press release or public announcement in written or electronic form upon such advance notice as is practicable under the circumstances for the purpose of allowing the notified party to review and comment upon such press release or public announcement. Under such circumstances, the releasing party shall not be obligated to delay making any such press release or public communication beyond the time when the same is required to be made. Neither party shall be required to seek the permission of the other party to repeat any information regarding the terms of this Agreement or any amendment hereto that has already been publicly disclosed by such party or by the other party in accordance with this Section 7.6(a); provided that such information remains accurate as of such time and provided the frequency and form of such disclosure are reasonable.
(b) Each party may make public statements regarding this Agreement in response to questions by the press, analysts, investors or those attending industry conferences or financial analyst calls, provided that any such public statement or press release: (i) is not inconsistent with prior public disclosures or public statements made in accordance with Section 7.6(a) or as permitted by Section 7.4; and (ii) does not reveal (A) information regarding the terms of this Agreement that have not previously been disclosed in accordance with Section 7.6(a) or as permitted by Section 7.4 or (B) non‑public information about the other party.
(c) The parties shall coordinate in advance with each other in connection with the filing of this Agreement (including redaction of certain provisions of this Agreement) with the SEC or other governmental agency or any stock exchange on which securities issued by a party or its Affiliate are traded, and neither party shall make any such filing unless the parties have mutually agreed upon the provisions to be redacted (such agreement not to be unreasonably withheld). The party filing this Agreement shall use reasonable efforts to seek and obtain confidential treatment for the provisions of this Agreement that the parties mutually agree to redact from such filing; provided that such party shall retain ultimate discretion to disclose such information to the SEC or any stock exchange or other governmental agency (as the case may be) as such party determines, based on advice of legal counsel, is required to be so disclosed. Except as expressly set forth in this Article 7, neither party (or its Affiliates) shall be obligated to consult with or obtain approval from the other party with respect to any filings with the SEC or any stock exchange or other governmental agency where such filings do not disclose Confidential Information of the other party.
7.8 Scientific Publications. Each party recognizes that publications regarding results of clinical and non-clinical studies carried out under this Agreement and other information regarding Licensed Products (including Expanded Licensed Product), including oral or poster presentations and abstracts (each of the foregoing, a “Publication”), may be beneficial to both parties provided such Publications are subject to reasonable controls to protect Confidential Information. Accordingly, a party shall have the right to review and comment on any Publication prior to submission by the other party. Before a Publication is submitted for publication or disclosure (other than oral presentation materials and abstracts, which are addressed below), the party proposing publication shall deliver a complete copy to the other party at least 21 days prior to submitting the material to a publisher or initiating such other disclosure, and such other party shall review any such material and give its comments to the party proposing publication within 10 days of the delivery of such material to such other party. With respect to oral presentation materials and abstracts, the party proposing publication shall deliver a complete copy to the other party at least 14 days prior to the anticipated date of the presentation, and such other party shall make reasonable efforts to expedite review of such materials and abstracts, and shall return such items as soon as practicable to the party proposing publication with appropriate comments, if any, but in no event later than 10 days from the date of delivery to the non-publishing party. The party seeking to publish or present shall consider in good faith any comments thereto provided by the other party and shall comply with the other party’s request to remove any and all of such other party’s Confidential Information in any such material. In addition, the party seeking to publish or present agrees to delay any submission for publication or other public disclosure for a period of up to an additional 30 days, in the event that the other party can demonstrate a reasonable need for such delay for the purpose of preparing and filing appropriate patent applications. If the other party fails to provide its comments to the party seeking to publish or present within the applicable 10-day period, such other party shall be deemed not to have any comments, and the party seeking to publish or present shall be free to submit for publication or present in accordance with this Section 7.6 after the 21-day period or 14-day period, as applicable, has elapsed (but without waiver of, or prejudice to, any liability of the publishing party for breach of its obligations pursuant to this Article 7).
8. | INTELLECTUAL PROPERTY |
8.1 Ownership. As between the parties, Cidara is and shall at all times be the sole and exclusive owner of all right, title and interest in and to the Cidara Technology. Inventorship of Inventions shall be determined in accordance with United States patent laws. Cidara shall solely own all Cidara Inventions. Mundipharma shall solely own all Mundipharma Inventions. The parties shall jointly own all Joint Inventions. Subject to the terms and conditions of this Agreement, and except to the extent that a party has granted the other party an exclusive license under such party’s joint ownership interest in Joint Inventions and Joint Patents, each party shall have the right to practice, and to grant licenses under, such party’s own joint ownership interest in Joint Inventions and Joint Patents without the other party’s consent, and shall have no duty to account to the other party for such practice or license, and each party hereby waives any right it may have under the laws of any country to require such consent or accounting.
8.2 Patent Prosecution and Maintenance. For purposes of this Section 8.2, the terms “prosecution” and “maintenance” (including variations such as “prosecute” and “maintain”) shall mean, with respect to a Patent, the preparation, filing, prosecution (including conducting all correspondence and interactions with any patent office and seeking, conducting and defending all any interferences, inter partes reviews, reissue proceedings, reexaminations, and oppositions and similar proceedings) and maintenance (including payment of any patent annuity fees) of such Patent, as well as re-examinations, reissues, appeals, post grant reviews (PGR), inter partes reviews (IPR) and requests for patent term adjustments, patent term extensions, supplementary protection certificates, or their equivalents with respect to such Patent, together with […***…]. For clarification, “prosecution” and “maintenance” (including variations such as “prosecute” and “maintain”) shall exclude any enforcement action with respect to a Patent, unless such enforcement action[…***…].
(a) Cidara Patents.
(i) Cidara Patents Other Than Cidara General Manufacturing/Formulation Patents.
(1) Cidara shall use Commercially Reasonable Efforts to prosecute and maintain the Patents derived from (x) international application […***…] and/or claiming priority from […***…] and (y) from international application […***…] and/or claiming priority from […***…], using counsel of its own choice, […***…]; provided, however, that the foregoing shall not be construed to prohibit ordinary course prosecution actions, including amending, or agreeing to amend, the scope of a claim of a pending patent application within the foregoing, or filing a terminal disclaimer with respect to or abandoning a claim of a pending patent application within the foregoing in favor of a related claim contained in another patent application filed by Cidara, subject to the remainder of this Section 8.2(a)(i).
(2) With respect to other Cidara Patents (excluding Patents referenced in Section 8.2(a)(i)(1) and the Cidara General Manufacturing/Formulation Patents), Cidara shall have the first right, but not the obligation, to prosecute and maintain in the Mundipharma Territory, using counsel of its own choice, […***…].
(3) Cidara shall keep Mundipharma reasonably informed of progress with regard to the prosecution and maintenance of all Cidara Patents (excluding the Cidara General Manufacturing/Formulation Patents) in the Mundipharma Territory, including any requests for patent term adjustments, patent term extensions, supplementary protection certificates or their equivalents. In addition, Cidara shall promptly provide Mundipharma with drafts of all proposed substantive filings and correspondence to any patent authority to the extent related to such Cidara Patents in the Mundipharma Territory for Mundipharma’s review and comment prior to the submission of such proposed filings and correspondence. Cidara shall consider in good faith Mundipharma’s comments related to such Cidara Patents prior to submitting such filings and correspondence, provided that Mundipharma provides such comments within 30 days (or a shorter period reasonably designated by Cidara if 30 days is not practicable given the filing deadline) of receiving the draft filings and correspondence from Cidara.
(4) In the event that Cidara seeks to abandon or cease the prosecution or maintenance of any Cidara Patent covered by Section 8.2(a)(i)(2) in the Mundipharma Territory (without initiation of the prosecution and maintenance of a substitution therefor), Cidara shall provide reasonable prior written notice to Mundipharma of such intention to abandon or cease such prosecution or maintenance (which notice shall be given no later than 30 days prior to the next deadline for any action that must be taken with respect to any such Cidara Patent in the relevant patent office in the Mundipharma Territory). […***…].
(ii) Cidara General Manufacturing/Formulation Patents. Cidara shall have the sole right, but not the obligation, to prosecute and maintain the Cidara General Manufacturing/Formulation Patents in the Mundipharma Territory […***…].
(b) Joint Patents. Cidara shall have the first right, but not the obligation, to control and manage the prosecution and maintenance of all Joint Patents in the Cidara Territory and the Mundipharma Territory, […***…]. Cidara shall consult with Mundipharma as to the prosecution and maintenance of Joint Patents reasonably prior to any deadline or action with any patent office, and shall furnish to Mundipharma copies of all relevant drafts and documents reasonably in advance of such consultation. Cidara shall keep Mundipharma reasonably informed of progress with regard to the prosecution and maintenance of Joint Patents and shall provide to Mundipharma copies of all material patent office submissions within a reasonable amount of time following submission thereof by Cidara. In the event that Cidara desires to abandon or cease the prosecution or maintenance of any Joint Patent in any country (without initiation of the prosecution and maintenance of a substitution therefor), Cidara shall provide reasonable prior written notice to Mundipharma of such intention to abandon (which notice shall, to the extent possible, be given no later than 30 days prior to the next deadline for any action that must be taken with respect to any such Joint Patent in the relevant patent office). In such case, at Mundipharma’s sole discretion, upon written notice to Cidara from Mundipharma, Mundipharma may elect to continue the prosecution and maintenance of any such Joint Patent, […***…].
8.3 Cooperation of the Parties. Each party agrees to cooperate fully in the preparation, filing, prosecution and maintenance of the Cidara Patents pursuant to Section 8.2. Such cooperation includes, but is not limited to: (a) executing all papers and instruments, or requiring its employees or contractors, to execute such papers and instruments, so as to effectuate the ownership of Inventions set forth in Section 8.1, and Patents claiming or disclosing such Inventions, and to enable the other party to apply for and to prosecute patent applications in any country as permitted by Section 8.2, and (b) promptly informing the other party of any matters coming to such party’s attention that may affect the prosecution and maintenance of any such patent applications.
8.4 Third Party Infringement. Each party shall notify the other party in writing within 10 Business Days (except as expressly set forth below) of becoming aware of any alleged or threatened infringement by a Third Party of any Cidara Patent or Joint Patent (“Infringement”), including (x) any such alleged or threatened Infringement on account of […***…] and (y) […***…] ((x) and (y), collectively, “Competitive Infringement”); provided, however, that each party shall notify the other party of any […***…] regarding any Cidara Patent or Joint Patent that it receives, and such party shall provide the other party with a copy of such[…***…], within five (5) Business Days of receipt.
(a) Cidara Patents.
(i) Cidara Patents. […***…] shall have the first right, but not the obligation, to bring and control any action or proceeding with respect to Competitive Infringement in the Field in the […***…] of any Cidara Patent[…***…], […***…], and […***…] shall have the right, […***…], to be represented in any such action […***…]. […***…] fails to bring any such action or proceeding within (i) […***…] following the notice of alleged infringement, or (ii) […***…] before the time limit, if any, set forth in the appropriate laws and regulations for the filing of such actions, whichever comes first, then […***…] shall have the right to bring and control any such action, […***…], and […***…] shall have the right, […***…], to be represented in any such action […***…]. […***…] shall have the sole right, but not the obligation, to bring and control any action or proceeding with respect to Infringement of any such Cidara Patent, other than Competitive Infringement in the Field […***…]. Each party shall keep the other party reasonably informed of the status and progress of any action or proceeding brought by such party with respect to Competitive Infringement in the Field […***…] of any Cidara Patent[…***…], and shall provide the other party with drafts of all proposed substantive filings in such action or proceeding reasonably in advance of making such filings and consider the other party’s comments on such filings in good faith.
(ii) […***…]. […***…] shall have the sole right, but not the obligation, to bring and control any action or proceeding with respect to Infringement of any[…***…], […***…]. With respect to any action or proceeding with respect to Competitive Infringement in the Field in the Mundipharma Territory of any […***…] shall (x) keep […***…] reasonably informed of the status and progress of any such action or proceeding, but […***…] shall not have the right to be represented, or otherwise participate, in such action or proceeding without the prior written consent of […***…], which […***…] may withhold in its sole discretion, and (y) to the extent reasonably practicable, provide […***…] with drafts of substantive filings in such action or proceeding reasonably in advance of making such filings and consider […***…] comments on such filings in good faith.
(b) Joint Patents. […***…] shall have the first right, but not the obligation, to bring and control any action or proceeding to enforce any Joint Patent with respect to Competitive Infringement in the Field […***…], […***…], and […***…] shall have the right, […***…], to be represented in any such action […***…]. […***…] shall have the first right, but not the obligation, to bring and control any action or proceeding to enforce any Joint Patent with respect to Competitive Infringement in the Field in the […***…] and by counsel of its own choice, and […***…] shall have the right[…***…], to be represented in any such action[…***…]. If the party having the first right to bring and control any action or proceeding to enforce any Joint Patent under this Section 8.4(b) (the “First Party”) fails to bring and control any such action or proceeding within (i) […***…] following the notice of alleged infringement, or (ii) […***…], whichever comes first, then the other party shall have the right to bring and control any such action, […***…], and the First Party shall have the right, […***…], to be represented in any such action[…***…]. In the case of Infringement of a Joint Patent, other than Competitive Infringement, the parties shall mutually agree in good faith on a case-by-case basis whether to jointly bring and control any action or proceeding to enforce such Joint Patent, or whether one party will bring and control any action or proceeding to enforce such Joint Patent, and, in each case, how the costs and expenses of such action or proceeding, and any recovery from such action or proceeding, will be allocated between the parties.
(c) Cooperation. In the event a party brings an infringement action in accordance with this Section 8.4 (such party, the “Enforcing Party”), the other party shall cooperate fully, including, if required to bring such action, the furnishing of a power of attorney or being named as a party. The Enforcing Party shall not […***…].
(d) Recovery. Except as otherwise agreed by the parties in connection with a cost-sharing arrangement, any recovery as a result of any action or proceeding pursuant to Section 8.4(a)(i), 8.4(a)(ii) or 8.4(b), whether by way of settlement or otherwise, shall first[…***…], and any remainder of the recovery after reimbursement of the litigation costs and expenses of the parties (“Remainder”), […***…]; provided, however, that:
(i) any Remainder of a recovery realized by […***…] as a result of any action brought and controlled by […***…] pursuant to Section 8.4(a)(i) that is specifically attributable to Competitive Infringement in the Field […***…] of a Cidara Patent shall be allocated as follows: (A) […***…],
(1) if such damages are assessed either on the basis of the […***…], or as […***…], in either case, it shall be […***…]; or
(2) if the recovery is assessed on the basis of […***…];
and (B) to the extent it represents […***…];
(ii) any Remainder of a recovery realized by […***…] as a result of any action brought and controlled by […***…] pursuant to Section 8.4(a)(ii) that is specifically attributable to Competitive Infringement in the Field […***…] of any […***…], shall be allocated as follows: (A) to the extent it represents […***…] in the Field […***…], […***…]% to Cidara and […***…]% to Mundipharma; and (B) to the extent it represents […***…] for Competitive Infringement of such […***…] in the Field […***…], […***…]% to […***…] and […***…]% to […***…]. For clarity, […***…] shall not be entitled to any portion of a recovery realized by […***…] as a result of […***…] pursuant to Section 8.4(a)(ii) that is specifically attributable to Infringement, […***…], in the Field […***…] of any […***…]; and
(iii) any Remainder of a recovery realized by […***…] as a result of any action brought and controlled by […***…] pursuant to Section 8.4(b) that is specifically attributable to Competitive Infringement in the Field […***…] of any Joint Patent shall be allocated as follows: (A) to the extent it represents […***…],
(1) if such damages are assessed either on the basis of the […***…], or as […***…], in either case, it shall be […***…]; or
(2) if the recovery is assessed on the basis of […***…];
and (B) to the extent it represents […***…].
8.5 Infringement of Third Party Rights. Each party shall promptly notify the other in writing of any allegation by a Third Party that the manufacture, use, importation, offer for sale or sale of Product infringes or may infringe the intellectual property rights of such Third Party. Cidara shall have the sole right to control any defense of any such claim involving alleged
8.6
infringement of Third Party rights by Cidara’s activities at its own expense and by counsel of its own choice. Mundipharma shall have the sole right to control any defense of any such claim involving alleged infringement of Third Party rights by Mundipharma’s activities at its own expense and by counsel of its own choice. Neither party shall […***…].
8.7 Patent Marking. Mundipharma shall xxxx (or cause to be marked) Licensed Product marketed and sold hereunder with appropriate Cidara Patent numbers or indicia to the extent required by Applicable Laws.
8.8 European United Patent Court. In the event that the Unitary Patent Court (“UPC”) enters into force, Cidara shall (a) at least ten (10) days before any necessary decision, provide Mundipharma with a proposal on whether to allow some, or all, of the Cidara Patents in Europe to become automatically subject to the jurisdiction of the UPC; or whether to opt them out. In response to this proposal, Mundipharma and its patent counsel may provide an alternative proposal for consideration. This proposal shall be taken into consideration in good faith by Cidara and its patent counsel, and each party shall cooperate with and assist in all reasonable respects and provide the other party all reasonable assistance and cooperation in connection with the decisions on whether Cidara Patents should be opted out or allowed to remain in the UPC. In the event that a decision is taken to opt-out one or more of such Cidara Patents in Europe, Cidara, at its sole expense and acting through patent counsel of its choice, shall be solely responsible for preparing and registering the opt-outs.
8.9 Trademarks.
(a) Licensed Product Trademark. Mundipharma shall have the right to select the Licensed Product-specific trademark for use in connection with the marketing and sale of Licensed Product in the Mundipharma Territory (the “Licensed Product Trademark”), which may, but need not, be the same as the Licensed Product-specific trademark used in connection with the marketing and sale of Licensed Product in the Cidara Territory (the “Cidara Product Trademark”). Mundipharma shall consider in good faith Cidara’s suggestions regarding the selection of the Licensed Product Trademark but shall retain ultimate discretion as to such selection.
(i) Use of Cidara Product Trademark. In the event that Mundipharma elects to use the Cidara Product Trademark in connection with the marketing and sale of Licensed Product in the Mundipharma Territory, then, effective upon such selection and subject to the terms and conditions of this Agreement, Cidara hereby grants to Mundipharma during the Term an exclusive, royalty-free license, with the right to sublicense solely in conjunction with the grant of a permitted Sublicense under Section 2.2, to use the Cidara Product Trademark in connection with the marketing and sale of Licensed Product in the Mundipharma Territory in accordance with this Agreement. During the Term, Mundipharma shall not use (or license to an Affiliate or Third Party) the Cidara Product Trademark in connection with any product other than Licensed Product, nor shall Mundipharma use (or cause or permit any Affiliate or Sublicensee to use) the Cidara Product Trademark outside of the Mundipharma Territory. Mundipharma shall be responsible for the failure by its Affiliates and Sublicensees to comply with this Section 8.8, including all relevant restrictions, limitations and obligations. Mundipharma shall obtain Cidara’s approval prior to the first use of the Cidara Product Trademark in any Licensed Product labeling, packaging or marketing materials, such approval not to be unreasonably withheld, conditioned or delayed if the Cidara Product Trademark is used in a manner that is consistent with Cidara’s reasonable usage guidelines for the Cidara Product Trademark in the Cidara Territory. Cidara shall own and retain all right, title and interest in and to the Cidara Product Trademark, and all goodwill associated with or attached to the Cidara Product Trademark arising out of the use thereof by Mundipharma, its Affiliates and Sublicensees shall vest in and inure to the benefit of Cidara. Mundipharma agrees not to contest, oppose or challenge Cidara’s ownership of the Cidara Product Trademarks. Mundipharma agrees not to do or suffer to be done, at any time, any act or thing that will in any way impair Cidara’s ownership of or rights in and to the Cidara Product Trademark or any registration thereof or that may depreciate the value of the Cidara Product Trademark. Mundipharma agrees that in using the Cidara Product Trademark upon any Licensed Product packaging, labeling, advertising or promotional materials, it shall not represent in any way that it has any right or title to the ownership of the Cidara Product Trademark or the registration thereof. Mundipharma shall, at Cidara’s request and expense, assist Cidara in any action reasonably necessary or desirable to protect the Cidara Product Trademark. Mundipharma shall as soon as practicable notify Cidara of any apparent infringement by a Third Party of the Cidara Product Trademark. The parties agree that, in the event there is an actual Third Party opposition to Mundipharma’s use of a Cidara Product Trademark in the Mundipharma Territory, (x) Mundipharma may request that Cidara enter into one or more coexistence agreements with such Third Part(ies) in respect of trademarks that such Third Parties own and/or utilize in the Mundipharma Territory, and (y) Cidara shall consider such request in good faith and work cooperatively with Mundipharma to address the opposition to the extent reasonably practical which may include entering into an appropriate coexistence agreement.
(ii) Use of Other Licensed Product Trademark. If Mundipharma elects to use a Licensed Product Trademark other than the Cidara Product Trademark in connection with the marketing and sale of Licensed Product in the Mundipharma Territory, Mundipharma shall own all right, title and interest in and to such Licensed Product Trademark, and all goodwill associated with or attached to such Licensed Product Trademark arising out of the use thereof by Mundipharma, its Affiliates and Sublicensees shall vest in and inure to the benefit of Mundipharma.
(b) Prosecution. Cidara shall control the prosecution of, and, if Mundipharma elects to use the Cidara Product Trademark as the Licensed Product Trademark, use Commercially Reasonable Efforts to maintain, the Cidara Product Trademark. Unless Mundipharma elects to use the Cidara Product Trademark as the Licensed Product Trademark, Mundipharma shall control the prosecution of, and use Commercially Reasonable Efforts to maintain, the Licensed Product Trademark at its expense.
(c) Enforcement. In the event that Mundipharma elects to use the […***…], each party shall promptly notify the other party in writing upon becoming aware of any infringement of the […***…] in the Mundipharma Territory. […***…] shall control the enforcement of the […***…] in the […***…] at its expense. If […***…] uses a […***…] […***…] in the […***…], […***…] shall control the enforcement of the […***…] in the […***…] at its expense.
8.10 Domain Names. Mundipharma may, at its cost, register as domain names the Cidara Product Trademark or Licensed Product Trademark in any country in the Mundipharma Territory using any available generic top-level domain or country-code top-level domain.
9. | REPRESENTATIONS AND WARRANTIES |
9.1 Mutual Representations and Warranties. Each party represents and warrants to the other party, as of the Effective Date, as follows:
(a) Corporate Power, Authority and Binding Agreement. (i) It is duly organized and validly existing under the laws of its jurisdiction of incorporation or formation, and has full corporate or other power and authority to enter into this Agreement and to carry out the provisions hereof; (ii) it is duly authorized to execute and deliver this Agreement and to perform its obligations hereunder, and the person or persons executing this Agreement on its behalf has been duly authorized to do so by all requisite corporate or partnership action; and (iii) this Agreement has been duly executed and delivered on behalf of such party and constitutes a legal, valid and binding obligation upon it, enforceable in accordance with its terms, subject to enforcement of remedies under applicable bankruptcy, insolvency, reorganization, moratorium or similar laws affecting generally the enforcement of creditors’ rights and subject to a court’s discretionary authority with respect to the granting of a decree ordering specific performance or other equitable remedies.
(b) No Conflict. The execution and delivery of this Agreement, the performance of such party’s obligations hereunder, and the licenses to be granted by such party pursuant to this Agreement do not and will not: (i) conflict with any agreement, instrument or understanding, oral or written, to which it or any of its Affiliates is a party or by which it or they may be bound; (ii) violate any Applicable Law or regulation of any court, governmental body or administrative or other agency having jurisdiction over it, existing as of the Effective Date; or (iii) conflict with or violate the certificate of incorporation, by-laws or other organizational documents of such party.
(c) No Consents. No authorization, consent, approval of a Third Party, nor any license, permit, exemption of, or filing or registration with, or notification to, any court or governmental authority is or will be necessary for the (i) valid execution, delivery or performance of this Agreement by such party; or (ii) the consummation by such party of the transactions contemplated hereby; except, in the case of Cidara, to the extent provided in the Cidara Loan and Security Agreement.
(d) Other Rights. Neither it, nor any of its Affiliates, is party to or otherwise bound by any contract or agreement, oral or written, that will result in any other person obtaining any interest in, or that would give to any other person any right to assert any claim in or with respect to, any of such party’s rights under this Agreement; except, in the case of Cidara, to the extent provided in the Cidara Loan and Security Agreement.
9.2 Cidara Representations and Warranties. Cidara hereby represents and warrants to Mundipharma, on behalf of itself and its Affiliates, as of the Effective Date, as follows:
(a) Patent Rights. Exhibit A attached hereto contains a true and complete list of the existing Patents as of the Effective Date that are Controlled by Cidara and/or any of its Affiliates and that are necessary or useful for (i) the development, registration, use or commercialization of Licensed Product in the Mundipharma Territory or (ii) the manufacture of Licensed Product worldwide (collectively, the “Existing Patents”), but, for clarity, Exhibit A does not include any patent application that has been abandoned, finally rejected or expired.
(b) Ownership of Existing Patents. Except to the extent provided in the Cidara Loan and Security Agreement, Cidara is the sole owner of the Existing Patents and has sufficient legal and beneficial title in and to the Existing Patents, free and clear of any mortgages, pledges, liens, security interests, charges, and encumbrances that would conflict with or derogate from the licenses, options and other rights granted to Mundipharma hereunder. Cidara has not granted any Third Party any license, option or right under or with respect to the Existing Patents that would conflict with or derogate from the licenses, options and other rights granted to Mundipharma hereunder.
(c) Compliance with Laws and Payment of Fees. Cidara has complied with all Applicable Laws in connection with the prosecution of the Existing Patents (including the duty of candour), and all material renewal and maintenance fees due with respect to the prosecution and maintenance of the Existing Patents have been paid.
(d) Cidara General Manufacturing/Formulation Patents. None of the Existing Patents are Cidara General Manufacturing/Formulation Patents.
(e) Intellectual Property Rights. The Cidara Technology includes all intellectual property rights Controlled by Cidara which is reasonably necessary or useful for the development, registration, manufacture, use and commercialization of Licensed Product as contemplated by the terms of this Agreement.
(f) Ownership of Existing Data. Cidara Controls the Data existing at the Effective Date that has arisen from the GLP Studies and clinical trials of the Licensed Product conducted by or on behalf of Cidara or any of its Affiliates, free and clear of any mortgages, pledges, liens, security interests, charges, and encumbrances that would conflict with or derogate from the licenses, options and other rights granted to Mundipharma hereunder. Cidara has not granted any Third Party any license, option or right under or with respect to the Data existing at the Effective Date that has arisen from the GLP Studies and clinical trials of the Licensed Product conducted by or on behalf of Cidara or any of its Affiliates that would conflict with or derogate from the licenses, options and other rights granted to Mundipharma hereunder.
(g) No Grant of Rights. Neither Cidara nor any of its Affiliates has granted to any Third Party or Affiliate any rights (including any license, or option or other right to obtain a license), to develop or commercialize Product in the Field in the Mundipharma Territory, except that Cidara has granted rights to the NIH to conduct the NIH Trial and to Third Party service providers to develop Product in the Field in the Mundipharma Territory on behalf of Cidara.
(h) Non-Infringement of Third Party Rights. To Cidara’s Knowledge, neither the conduct of the Lead Indication Trials nor the manufacture of the Lead Indication Product (as manufactured on behalf of Cidara as of the Effective Date) infringes, or would infringe, the Patents of any Third Party or misappropriates the proprietary information of any Third Party.
(i) Intellectual Property Challenge. Neither Cidara nor any of its Affiliates has received any written notice from any Third Party seeking to invalidate or otherwise challenge the ownership, scope, validity and/or enforceability of the claims of the Existing Patents and, to Cidara’s Knowledge, there is no such challenge pending or threatened.
(j) Notice of Infringement. Neither Cidara nor any of its Affiliates has received any written notice from any Third Party claiming that the development, manufacture, use, sale, offer for sale, export or import of Compound or Product infringes, misappropriates or would infringe or misappropriate the Patent or other intellectual property rights of any Third Party.
(k) No Unauthorized Use and Non-Infringement of Rights. To Cidara’s Knowledge, no Third Party is infringing or misappropriating, or has infringed or misappropriated, any of the Cidara Technology.
(l) Proceedings and Investigations. Other than patent office actions and proceedings related to the Existing Patents and the actions and proceedings of Regulatory Authorities related to the clinical development of Product by Cidara, neither Cidara nor its Affiliates is a party to any adverse action, suit, investigation or proceeding relating to the Cidara Technology, Compound or Product, and neither Cidara nor its Affiliates have received written notice of any pending or threatened adverse action, suit, investigation or proceeding related to the Cidara Technology, Compound or Product.
(m) Accuracy of Information. All tangible or recorded information and data (including Data) provided by or on behalf of Cidara to Mundipharma or its Affiliates related to Product on or before the Effective Date in contemplation of this Agreement was and is true, accurate and complete in all material respects, and Cidara has not failed to disclose, or failed to cause to be disclosed, any such information or data (including Data) related to Compound or Product in its possession and Control that would cause the information and data that has been disclosed to be misleading in any material respect.
(n) Material Contracts. Cidara has disclosed to Mundipharma all material contracts to which Cidara or any of its Affiliates is or was a party, (i) pursuant to which Cidara or any of its Affiliates acquired or obtained Control of any Existing Patent or any invention claimed by any Existing Patent, (ii) pursuant to which Data was generated; (iii) pursuant to which any Third Party has conducted, or is conducting, any GLP Study or clinical trial of Compound or Licensed Product on behalf of Cidara or any of its Affiliates, or (iv) pursuant to which any Third Party has conducted, or is conducting, any CMC development activities with respect to Compound or Licensed Product on behalf of Cidara or any of its Affiliates, or has manufactured and supplied to Cidara or any of its Affiliates, or is obligated to manufacture and supply to Cidara or any of its Affiliates, any GLP-compliant or GMP-compliant Compound or Licensed Product.
(o) Employee, Consultant and Contractor Agreements. All current and former employees, paid consultants and Third Party contractors of Cidara who are or were substantively involved in the research, development or manufacture of Compound or Product (i) have executed written contracts containing, or are otherwise bound by, customary obligations of confidentiality and non‑use with respect to Cidara Technology, and (ii) have assigned to Cidara all right, title and interest in and to all inventions, Information and Data made or generated in the course of research, development or manufacture of Compound or Product.
(p) Third Party Confidentiality. No Third Party has received from Cidara or any of its Affiliates, or, to its Knowledge, its Representatives (that are not directors, officers or employees of Cidara or any of its Affiliates), any Data resulting from GLP Studies or clinical trials of Licensed Product that is within Cidara Know-How, or any other material Cidara Know-How, that, in each case, is Confidential Information of Cidara as of the Effective Date, which is not subject to customary obligations of confidentiality owed to Cidara or such Affiliate.
(q) Communications with Regulatory Authorities. Cidara: (i) has provided or made available all relevant documents and written communications and materials in its and its Affiliates’ possession or Control from and to any Regulatory Authority related to the Product; (ii) as of the Effective Date, Cidara and its Affiliates have not received any oral or written communication (including any notice of inspection, deficiency letter, warning letter or similar notice) from any Regulatory Authority alleging that the development of Product by or on behalf of Cidara and its Affiliates is not currently materially in compliance with any and all applicable laws or regulations implemented by any Regulatory Authority; and (iii) Cidara and its Affiliates have not made, with respect to the Product, any untrue statement of material fact to any Regulatory Authority, or failed to disclose a material fact required to be disclosed to such Regulatory Authority.
(r) Safety and Efficacy. Cidara has (i) allowed Mundipharma access to all material information in Cidara or its Affiliates’ possession and Control concerning the safety and efficacy of Compound or Licensed Product; (ii) informed Mundipharma of all drug-related adverse reactions relating to the Product or its use of which Cidara and its Affiliates are aware; and (iii) carried out GLP Studies of Compound and Licensed Product in accordance with GLP and clinical trials of Licensed Product in accordance with GCP.
(s) Debarment. Neither Cidara nor any of its Affiliates is or has been debarred or suspended under 21 U.S.C. §335(a) or §335(b) or any foreign equivalent thereof, or is the subject of a conviction described in such section or any foreign equivalent thereof.
(t) Anti-Corruption Laws. Cidara has not: (i) taken any action in violation of any Anti-Corruption Laws; or (ii) directly or indirectly through Affiliates or Third Parties, paid, promised or offered to pay, or authorized the payment of, any money or given any promise or offered to give, or authorized the giving of anything of value to a public official or Entity or other Person for purpose of obtaining or retaining business for or with, or directing business to, any Person, including to Cidara and its Affiliates, nor has Cidara or any of its Affiliates directly or indirectly promised, offered or provided any corrupt payment, gratuity, emolument, bribe, kickback, illicit gift or hospitality or other illegal or unethical benefit to a public official or entity or any other Person.
(u) Affiliates. Cidara has only two Affiliates, both of which are Cidara-Controlled Affiliates: (i) Cidara Therapeutics UK Limited; and (ii) Cidara Therapeutics (Ireland) Limited.
9.3 Mundipharma Representations and Warranties. Mundipharma represents and warrants to Cidara, as of the Effective Date, as follows:
(a) Debarment. Neither Mundipharma nor any of its Affiliates has been debarred or suspended under 21 U.S.C. §335(a) or §335(b) or any foreign equivalent thereof, or is the subject of a conviction described in such section or any foreign equivalent thereof.
(b) Anti-Corruption Laws. Neither Mundipharma nor any of its Affiliates has: (i) taken any action in violation of any Anti-Corruption Laws, or (ii) directly or indirectly through Affiliates or Third Parties, paid, promised or offered to pay, or authorized the payment of, any money or given any promise or offered to give, or authorized the giving of anything of value to a public official or Entity or other Person for purpose of obtaining or retaining business for or with, or directing business to, any Person, including to Mundipharma and its Affiliates, nor has Mundipharma or any of its Affiliates directly or indirectly promised, offered or provided any corrupt payment, gratuity, emolument, bribe, kickback, illicit gift or hospitality or other illegal or unethical benefit to a public official or entity or any other Person.
9.4 Mutual Covenants. In addition to any covenants made by it elsewhere in this Agreement, each party hereby covenants to the other party that:
(a) in the event that such party becomes aware that (i) it or any of its Affiliates has been debarred, suspended or is the subject of a conviction described in 21 U.S.C. §335(a) or §335(b) or any foreign equivalent thereof, or (ii) if any action, suit, claim, investigation, or legal or administrative proceeding is pending or, to its actual knowledge, is threatened, relating to such debarment, suspension or conviction, such party will immediately notify the other party in writing, and, to the extent such notice is referring to a such a debarment, suspension or conviction pursuant to clause (i), the other party may terminate this Agreement immediately upon written notice to the other party; provided, however, that if any of a party’s Affiliates, but not such party itself, is debarred, suspended or is the subject of a conviction described in 21 U.S.C. §335(a) or §335(b) or any foreign equivalent thereof, then such party may avoid termination of this Agreement by the other party by immediately terminating all rights, licenses and sublicenses (including any Sublicense) granted by such party to that Affiliate and notifying the other party thereof in writing;
(b) in the event that such party becomes aware that any Person that is performing activities hereunder on its behalf has been debarred, suspended or is the subject of a conviction described in 21 U.S.C. §335(a) or §335(b) or any foreign equivalent thereof, or if any action, suit, claim, investigation, or legal or administrative proceeding is pending or, to its actual knowledge, is threatened, relating to such debarment, suspension or conviction, such party will immediately notify the other party in writing and such party will cease, or cause its Affiliate to cease (as applicable), employing, contracting with, or retaining any such Person to perform any services relating to Product;
(c) neither such party nor any of its Affiliates will, directly or indirectly through Affiliates or Third Parties, pay, promise or offer to pay, or authorize the payment of, any money or give any promise or offer to give, or authorize the giving of anything of value to a public official or Entity or other Person for purpose of obtaining or retaining business for or with, or directing business to, any Person, including such party and its Affiliates, nor will such party or any of its Affiliates directly or indirectly promise, offer or provide any corrupt payment, gratuity, emolument, bribe, kickback, illicit gift or hospitality or other illegal or unethical benefit to a public official or entity or any other Person;
(d) neither such party nor any of its Affiliates (or any of their respective employees and contractors), in connection with the exercise of such party’s rights or performance of such party’s obligations under this Agreement, shall cause the other party to be in violation of Anti-Corruption Laws or Export Control Laws;
(e) such party shall immediately notify the other party if such party has any information that there is or is likely to be a violation of Anti-Corruption Laws or Export Control Laws in connection with the exercise of such party’s rights or performance of such party’s obligations under this Agreement; and
(f) each party shall undertake due diligence activities appropriate to its activities under this Agreement in accordance with applicable Anti-Corruption Laws and related guidance, including guidance issued by the U.S. Department of Justice Criminal Division (entitled “Evaluation of Corporate Compliance Programs”) as amended from time to time, concerning the Foreign Corrupt Practices Act (15 U.S.C. §§78dd-1, et. seq.), and issued by the U.K. Ministry of Justice concerning the UK Xxxxxxx Xxx 0000 as amended from time to time, such activities to include the conduct of appropriate due diligence in relation to Third Party contractors conducting any GLP Study or clinical trial on such party’s behalf (including, in the case of Cidara, those Third Party contractors conducting the Lead Indication Trials) and, without prejudice to each party’s obligation pursuant to clause (ii) of Section 9.1(b), shall collaborate with the other party to ensure such compliance.
Each party has the right, upon reasonable notice and at its sole expense, to conduct, or have conducted by an independent Third Party reasonably acceptable to the other party, no more than once every three years (except for cause), a reasonable and customary audit of the other party for the purposes of monitoring compliance with this Section 9.4, and the other party shall, subject to compliance with Applicable Laws, provide to such party any relevant documents reasonably requested by such party in relation thereto. Save in respect of such an audit for cause, the auditing party shall reimburse the audited party for reasonable and documented external costs and expenses incurred by the audited party in complying with the foregoing requirements.
9.5 Cidara Covenants.
(a) Third Party Licenses. During the Term, Cidara shall not grant any Third Party any license or other right with respect to any Compound, Product or Cidara Technology in derogation of the licenses and rights expressly granted to Mundipharma hereunder.
(b) Control of Cidara Technology. During the Term, neither Cidara nor any Cidara-Controlled Affiliate shall delegate or contract the performance of any activity under this Agreement that Cidara is responsible for performing to any Affiliate of Cidara that is not a Cidara-Controlled Affiliate, unless, in each case: (i) none of Mundipharma’s rights hereunder are diminished or otherwise adversely affected as a result of such delegation or contracting; (ii) each such Affiliate has made a present assignment in writing to Cidara or a Cidara-Controlled Affiliate of all right, title and interest in and to any and all Inventions and Data generated or made by such Affiliate in the course of performing the delegated or contracted activities, as necessary for Cidara to grant to Mundipharma the full scope of licenses and rights to such Inventions (including Patents claiming such Inventions) and Data expressly contemplated by this Agreement; and (iii) each such Affiliate undertakes in writing obligations of confidentiality and non‑use regarding Confidential Information which are at least as stringent as those undertaken by the parties pursuant to Article 7. In addition, during the Term, Cidara shall not transfer or assign, and shall not permit any Cidara-Controlled Affiliate to transfer or assign, any Cidara Technology or Cidara’s interest in any Joint Technology to any Affiliate of Cidara that is not a Cidara-Controlled Affiliate (except in conjunction with a permitted assignment by Cidara of this Agreement and all of Cidara’s rights and obligations hereunder in accordance with Section 13.7). Cidara further acknowledges and agrees that any permitted Affiliate transferee of any Cidara Technology or Cidara’s interest in any Joint Technology will take such Cidara Technology or Cidara’s interest in such Joint Technology or the applicable rights thereunder subject to the terms of this Agreement, including the License, and Cidara shall obtain such permitted Affiliate transferee’s written acknowledgment of, and agreement to, the foregoing.
(c) […***…]
(d) […***…]
(e) Existing Contracts. Cidara shall promptly notify Mundipharma of, and allow Mundipharma a reasonable opportunity to review and comment upon, any amendment, variation, modification, supplement or other change to any of the following existing agreements with Cidara’s CMOs (each, an “Existing CMO Agreement”) that is proposed before the date on which Cidara signs the CMO Supply Agreement contemplated by Section 4.18(c) with the applicable CMO, that would adversely affect or limit any representations or warranties with respect to anidulafungin, Compound or Licensed Product (as applicable), or any remedies (including indemnities, and exclusions and limitations of liability) for defective anidulafungin, Compound or Licensed Product (as applicable), manufactured by such CMO that are contained in such Existing CMO Agreement as of the Effective Date:
(i) Master Agreement for Pharmaceutical Development and Technology Transfer Services between Cidara and […***…] dated March 13, 2017;
(ii) Master Services Agreement between Cidara and […***…] dated June 17, 2015;
(iii) Master Services Agreement between Cidara and […***…] dated November 3, 2017; and
(iv) Master Services Agreement between Cidara, […***…] dated 2015.
(f) Compliance Declarations. As soon as reasonably practicable following the Effective Date, Cidara shall obtain from the CMOs of Cidara that synthesize starting material (anidulafungin) for use in the manufacture of the Compound to be used in Licensed Product, and provide to Mundipharma, reasonable documentary evidence that such CMOs are in compliance with applicable laws concerning the characterization and genealogy of the master cell bank used or intended to be used for the research, development and manufacture of Compound and Licensed Product, including the Nagoya Protocol on Access to Genetic Resources and the Fair and Equitable Sharing of Benefits Arising from their Utilization to the Convention on Biological Diversity. If Cidara is not able to obtain such evidence of compliance from such CMOs, Cidara shall promptly use Commercially Reasonable Efforts to procure that such CMOs take steps necessary to ensure such compliance and thereafter maintain adequate records in connection therewith, as may be required by Regulatory Authorities or other governmental authorities of competent jurisdiction.
9.6 Performance by Affiliates, Sublicensees and Third Party Contractors. The parties recognize that each party may perform some or all of its obligations or exercise some or all of its rights under this Agreement through one or more Affiliates, Third Party contractors, or, in the case of Mundipharma and subject to Section 2.2, Sublicensees, Marketing Partners and Distributors; provided, in each case, that (a) none of the other party’s rights hereunder are diminished or otherwise adversely affected as a result of such delegation or contracting, and (b) each such Third Party contractor, and, in the case of Mundipharma, Sublicensee, Marketing Partner and Distributor, undertakes in writing obligations of confidentiality and non‑use regarding Confidential Information which are at least as stringent as those undertaken by the parties pursuant to Article 7; and provided, further, that each such Third Party contractor of a party agrees in writing to assign to such party any and all Inventions and Data generated or made by such contractor in the course of performing the contracted activities, so that such party can comply with its obligations under this Agreement. Each party shall at all times be fully responsible for the performance of its Affiliates, Third Party contractors, and, in the case of Mundipharma, Sublicensees, Marketing Partners and Distributors.
9.7 Disclaimer. Except as expressly set forth in this Agreement, THE TECHNOLOGY AND INTELLECTUAL PROPERTY RIGHTS PROVIDED BY CIDARA AND MUNDIPHARMA HEREUNDER ARE PROVIDED “AS IS.” Except as expressly set forth in this Agreement, NEITHER PARTY MAKES ANY REPRESENTATION OR EXTENDS ANY WARRANTIES OF ANY KIND, AND EACH PARTY EXPRESSLY DISCLAIMS ANY AND ALL WARRANTIES OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING, WITHOUT LIMITATION, THE WARRANTIES OF DESIGN, MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE, NON-INFRINGEMENT OF THE INTELLECTUAL PROPERTY RIGHTS OF THIRD PARTIES, OR ARISING FROM A COURSE OF DEALING, USAGE OR TRADE PRACTICES.
9.8 Limitation of Liability. Except in the case of […***…], NEITHER PARTY SHALL BE ENTITLED TO RECOVER FROM THE OTHER PARTY ANY SPECIAL, INCIDENTAL, CONSEQUENTIAL OR PUNITIVE LOSSES OR DAMAGES IN CONNECTION WITH THIS AGREEMENT OR ANY LICENSE GRANTED HEREUNDER; provided, however, that this Section 9.8 shall not be construed to limit […***…], or any liability (whether in contract, tort or otherwise) which cannot by law be excluded or limited.
10. | TERM AND TERMINATION |
10.1 Term. The term of this Agreement (the “Term”) shall commence on the Effective Date and, unless earlier terminated pursuant to Section 9.4(a) or this Article 10, shall continue until the expiration of the last-to-expire Royalty Term for any Licensed Product.
10.2 Termination For Cause.
(a) Material Breach.
(i) A party shall have the right to terminate this Agreement before the end of the Term upon written notice to the other party if such other party is in material breach of this Agreement and has not cured such breach within 60 days (or 30 days with respect to any payment breach) after notice from the terminating party requesting cure of the breach. Any such termination shall become effective at the end of such 60-day (or 30-day with respect to any payment breach) period unless the breaching party has cured such breach prior to the end of such period. Any right to terminate under this Section 10.2(a) shall be stayed and the cure period tolled in the event that, during any cure period, the party alleged to have been in material breach shall have initiated dispute resolution in accordance with Article 12 with respect to the alleged breach, which stay and tolling shall continue until such dispute has been resolved in accordance with Article 12.
(ii) In the event of material breach of this Agreement by Cidara that is not cured within the applicable notice period set forth in Section 10.2(a)(i) or is not curable (in each case, including as determined or agreed pursuant to dispute resolution in accordance with Article 12), Mundipharma, at its sole discretion, may either:
(1) terminate this Agreement in accordance with Section 10.2(a)(i) (in addition to pursuing any remedy that may be available to Mundipharma at law or in equity as a result of Cidara’s breach of this Agreement); or
(2) elect (A) not to terminate this Agreement, (B) to retain the License, subject to all terms and conditions hereof, and (C) pursue any remedy that may be available to Mundipharma at law or in equity as a result of Cidara’s breach of this Agreement, without prejudice to Mundipharma’s right to terminate this Agreement at a later date pursuant to Section 10.2(a)(i) (for that uncured material breach or any other uncured material breach of this Agreement by Cidara) or pursuant to Section 10.3; provided, however, that to the extent that Development Milestone Payment 1 has not been fully credited against royalties payable by Mundipharma hereunder or otherwise repaid in full in accordance with Section 5.3, any remaining amount not already credited or repaid shall become immediately due and payable.
(b) Patent Challenge. Cidara shall have the right to terminate this Agreement immediately upon written notice to Mundipharma if Mundipharma or any of its Affiliates or Sublicensees, directly or indirectly through any Third Party, commences any interference or opposition proceeding with respect to, challenges the validity or enforceability of, or opposes any extension of or the grant of a supplementary protection certificate with respect to, any Cidara Patent.
10.3 Mundipharma Termination At Will. After the first anniversary of the Effective Date], Mundipharma shall have the right to terminate this Agreement on a country-by-country basis or in its entirety for any reason or no reason at any time upon 90 days’ prior written notice to Cidara; provided, however, that no such termination of this Agreement in any country of the Mundipharma Territory but not in all countries of the Mundipharma Territory shall relieve Mundipharma of its obligations under Sections 4.1 and 4.2, other than Mundipharma Territory-Specific obligations in such terminated country; and provided, further, that if […***…] terminates this Agreement in its entirety […***…], […***…] shall continue to be liable for […***…]. Within 10 days after delivery of written notice pursuant to this Section 10.3, the JSC shall convene to discuss transition planning, subject to Section 10.5(d).
10.4 Termination for Mundipharma Insolvency. Cidara shall have the right to terminate this Agreement upon written notice to Mundipharma […***…] if Mundipharma incurs an Insolvency Event.
10.5 Effect of Expiration or Termination.
(a) Expiration. Upon expiration (but not earlier termination) of this Agreement pursuant to Section 10.1: (i) the License shall automatically become fully-paid, royalty-free, irrevocable and perpetual; (ii) the Grant-Back License shall survive; and (iii) all other rights and obligations of the parties under this Agreement shall terminate, except as provided elsewhere in this Section 10.5 or in Section 10.6 or Section 10.7.
(b) Effects of Termination. In the event of any termination of this Agreement by either party in accordance with the terms of this Agreement, the following shall occur:
(i) The License shall automatically terminate and revert to Cidara;
(ii) As promptly as practicable (and in any event within 45 days) after termination, Mundipharma shall, subject to Section 10.5(c)(ii): (A) to the extent not previously provided to Cidara, deliver to Cidara true, correct and complete copies of all Product Filings in the Field in the Mundipharma Territory (in each case, whether held in the name of Mundipharma, its Affiliate or a Sublicensee), and disclose to Cidara all previously-undisclosed Mundipharma Know-How; (B) transfer or assign, or cause to be transferred or assigned, to Cidara or its designee (or to the extent not so assignable, take all reasonable actions to make available to Cidara or its designee the benefits of all Product Filings in the Field in the Mundipharma Territory (in each case, whether held in the name of Mundipharma, its Affiliate or a Sublicensee); and (C) take such other actions and execute such other instruments, assignments and documents as may be necessary to effect, evidence, register and record the transfer, assignment or other conveyance of such rights to Cidara;
(iii) Mundipharma shall, as directed by Cidara, either promptly wind-down any ongoing development activities with respect to Licensed Product in an orderly fashion or promptly transition such development activities to Cidara or its designee; in each case, with due regard for patient safety and in compliance with all Applicable Laws and international guidelines. In addition, Mundipharma shall, as directed by Cidara, assign to Cidara or its designee any or all clinical trial agreements with respect to such development activities for Licensed Product (or to the extent not so assignable, take all reasonable actions to make available to Cidara or its designee the benefits of such agreements);
(iv) Subject to Section 10.6, Mundipharma shall reasonably cooperate, at Cidara’s request, with Cidara and its designee(s) to facilitate a smooth, orderly and prompt transition of any or all ongoing manufacturing and commercialization activities with respect to Licensed Product to Cidara or its designee(s). In addition, Mundipharma shall, as directed by Cidara, assign to Cidara or its designee any or all agreements with Third Party contractors for the manufacture and/or commercialization of Licensed Product (or to the extent not so assignable, take all reasonable actions to make available to Cidara or its designee the benefits of such agreements);
(v) Except otherwise expressly provided in Section 10.5(d) in the case of termination by Mundipharma pursuant to Section 10.3, all Sublicenses granted by Mundipharma to any of its Affiliates shall automatically terminate upon such termination and be of no further force or effect; and any Sublicense granted by Mundipharma to a Third Party that was in effect immediately prior to such termination shall survive and shall be assigned by Mundipharma to Cidara such that such Sublicense becomes a direct license from Cidara to such Sublicensee on the same terms as those set forth in this Agreement, to the extent applicable to the scope of the Sublicense granted by Mundipharma to such Sublicensee; provided, however, that such Sublicense was granted in accordance with the terms of Section 2.2, and that such Sublicensee is in compliance with the terms of the Sublicense agreement (and did not cause Mundipharma to breach its obligations under this Agreement) and agrees to comply with all applicable terms of this Agreement to the extent applicable to the scope of the Sublicense granted by Mundipharma to such Sublicensee; and provided, further, that in no event shall Cidara be bound by obligations contained in any Sublicense agreement that extend beyond the obligations of Cidara set forth in this Agreement;
(vi) Cidara shall have the first right, but not the obligation, to Prosecute all Joint Patents throughout the world, at Cidara’s sole expense and by counsel selected by Cidara. In the event that, after the effective date of such termination, Cidara desires to abandon or cease Prosecution of any Joint Patent, Cidara shall provide written notice to Mundipharma of such intention to abandon promptly after Cidara makes such determination (which notice shall be given no later than 30 days prior to the next deadline for any action that must be taken with respect to such Joint Patent in the relevant patent office). In such case, Mundipharma shall have the right, in its discretion, exercisable upon written notice to Cidara, to assume responsibility for Prosecution of such Joint Patent, at its sole cost and expense and by counsel of its own choice. Each party will cooperate with the other party and provide the other party with reasonable assistance with such Prosecution activities with respect to Joint Patents;
(vii) Cidara shall have the first right, but not the obligation, to bring and control any action or proceeding to enforce any Joint Patent with respect to Competitive Infringement anywhere in the world, at its own expense and by counsel of its own choice. If Cidara fails to bring and control any such action or proceeding within (A) 120 days following the notice of alleged infringement, or (B) 30 days before the time limit, if any, set forth in the appropriate laws and regulations for the filing of such actions, whichever comes first, then Mundipharma shall have the right to bring and control any such action, at its own expense and by counsel of its own choice, and Cidara shall have the right, at its own expense, to be represented in any such action by counsel of its own choice. In the event a Party brings an infringement action in accordance with this paragraph, the other Party shall cooperate fully, including, if required to bring such action, the furnishing of a power of attorney or being named as a party. The Party that brings such infringement action shall not enter into any settlement or compromise of any action under this paragraph: (1) in a manner that would diminish the rights or interests of the other Party without the written consent of such other Party, which shall not be unreasonably withheld; or (2) that would impose any cost or liability on the other Party, or admit the invalidity or unenforceability of any Joint Patent, without such other Party’s prior written consent, which may be withheld in such other Party’s sole discretion. Except as otherwise agreed by the Parties in connection with a cost-sharing arrangement, any recovery as a result of any action or proceeding pursuant to paragraph, whether by way of settlement or otherwise, shall first be used first to reimburse the Party that brought and controlled such action or proceeding for its documented, out-of-pocket costs and expenses (including court, attorneys’ and professional fees) incurred in connection with such action or proceeding, and then to reimburse the other Party for its documented, out-of-pocket costs and expenses (including court, attorneys’ and professional fees) incurred in connection with such action or proceeding (to the extent not previously reimbursed by the Enforcing Party), and any remainder of the recovery after reimbursement of the litigation costs and expenses of the Parties, shall be retained by the Enforcing Party. In the case of infringement of a Joint Patent, other than Competitive Infringement, the Parties shall mutually agree in good faith on a case-by-case basis whether to jointly bring and control any action or proceeding to enforce such Joint Patent, or whether one Party will bring and control any action or proceeding to enforce such Joint Patent, and, in each case, how the costs and expenses of such action or proceeding, and any recovery from such action or proceeding, will be allocated between the Parties;
(viii) if Mundipharma was, prior to termination, manufacturing, or having manufactured on its behalf, any quantities of Licensed Product, then at Cidara’s request, until the earlier of (A) such time as Cidara has secured another source thereof that is able to meet Cidara’s quality and quantity requirements, and (B) […***…] months after such termination, Mundipharma shall use commercially reasonable efforts to supply, or cause to be supplied, to Cidara such quantities
(ix)
thereof as Cidara may reasonably require for the development and commercialization of Licensed Products in the Field; provided that Cidara shall use Commercially Reasonable Efforts to secure another source of supply as soon as reasonably practicable. Such material shall be provided at the transfer price paid by Cidara to Mundipharma immediately prior to the date of termination;
(x) Cidara shall remit payment to Mundipharma of the remaining portion of the Development Milestone Payment 1 that has not, as of the effective date of termination, been credited against royalties payable to Cidara by, or otherwise refunded by Cidara to, Mundipharma; and
(xi) All other rights and obligations of the parties under this Agreement shall terminate, except as provided in Section 10.3, elsewhere in this Section 10.5, and in Sections 10.6 and 10.7.
(c) Termination by Mundipharma Pursuant to Section 9.4(a) or Section 10.2(a). In addition to the effects of termination set forth in Section 10.5(b), in the event of termination of this Agreement by Mundipharma pursuant to Section 10.2(a) the following shall apply:
(i) The Grant-Back License shall survive such termination. Any expansion of the Grant-Back License (i.e., to obtain worldwide rights and/or full exclusivity with respect to Compound and Product) that occurred prior to such termination shall survive such termination in accordance with the terms mutually agreed by the parties in good faith prior to such termination, and any such expansion of the Grant-Back License that occurs after such termination shall be on a royalty-bearing basis and otherwise on commercially reasonable terms to be negotiated by the parties in good faith;
(ii) If Mundipharma or its Affiliate had generated any Expanded Licensed Product Clinical Efficacy Data with respect to any Expanded Licensed Product in respect of which the parties had not executed an Expanded Licensed Product Amendment prior to such termination, then Cidara must pay […***…] with respect thereto if Cidara wishes to use such Expanded Licensed Product Clinical Efficacy Data in support of any NDA, MAA or other Product Filing or Regulatory Approval for the applicable Expanded Licensed Product or in the commercialization of such Expanded Licensed Product anywhere in the world. Promptly upon written request by Cidara, Mundipharma shall deliver a written invoice to Cidara for […***…], and Cidara shall pay the invoiced amount within 30 days of invoice;
(iii) Mundipharma shall, upon Cidara’s request, grant Cidara a royalty-bearing license to all of Mundipharma’s and its Affiliates’ right, title and interest in and to the Licensed Product Trademarks (if other than the Cidara Licensed Product Trademark), including, in each case, all goodwill therein, on commercially reasonable terms to be negotiated in good faith; and
(iv) Mundipharma may dispose of all inventory of Licensed Product remaining in Mundipharma’s or its Affiliates’ possession as of the effective date of termination
(v)
for up to six (6) months after the effective date of termination, subject to Mundipharma’s royalty payment, Net Sales Milestone Payment and reporting obligations to Cidara under Section 5.6, Section 5.7 (subject to Sections 5.8 through 0) and Article 6, respectively;
(vi) Subject to Mundipharma’s rights under Section 10.5(e)(iv), Cidara shall have the right, but not the obligation, to purchase from Mundipharma any or all usable inventory of Licensed Product in Mundipharma’s or its Affiliates’ possession as of the date of termination at a supply price equal to […***…]. Any packaging, transport, insurance and other costs relating to delivery shall be at Cidara’s expense;
(vii) In the event Mundipharma terminates this Agreement pursuant to Section 10.2(a) prior to the […***…] anniversary of the First Commercial Sale of Licensed Product in the Mundipharma Territory, then Mundipharma shall be entitled to recoup the portion of Global Development Costs paid to Cidara pursuant to Section 5.2 as of the effective date of termination in the form of a reverse royalty which shall be payable by Cidara as follows: Cidara shall pay Mundipharma a quarterly royalty in an amount equal to […***…]% of Net Sales of Licensed Product sold by Cidara, its Affiliates and/or licensees in the Mundipharma Territory; provided that Cidara’s obligation under this subsection shall terminate upon the first to occur of i) Mundipharma recouping all such Global Development Costs paid to Cidara pursuant to Section 5.2 as of the effective date of termination, and ii) expiration of the […***…] anniversary of the First Commercial Sale of Licensed Product in the Mundipharma Territory
(viii) Until the […***…] anniversary of the termination date, Cidara and its Affiliates shall not directly commercialize Licensed Product in the Mundipharma Territory, including with the appointment of a Distributor (which definition shall apply mutatis mutandis to Cidara). For clarity, during such […***…] year period Cidara and its Affiliates shall have the right to appoint a Third Party, or grant a Third Party a license, to market, promote and sell Licensed Product in the Mundipharma Territory; provided, however, that such Third Party is not a Distributor or Marketing Partner (which definitions shall apply mutatis mutandis to Cidara).
(d) Termination by Mundipharma Pursuant to Section 10.3 or by Cidara Pursuant to Section 10.4. In addition to the effects of termination set forth in Section 10.5(b), in the event of termination of this Agreement by Mundipharma pursuant to Section 10.3 or by Cidara pursuant to Section 10.4, the following provisions shall apply:
(i) The Grant-Back License shall survive such termination;
(ii) All Sublicenses granted by Mundipharma to any of its Affiliates shall automatically terminate and be of no further force or effect (or, in the case of termination on a country-by-country basis, shall automatically terminate and be of no further force or effect in the country(ies) in which Mundipharma has terminated this Agreement but continue in full force and effect in accordance with the terms of this Agreement and the applicable Sublicense agreement in the country(ies) in which Mundipharma has not terminated this Agreement) upon such termination; and any Sublicense granted by Mundipharma to a Third Party that was in effect immediately prior to such termination shall automatically terminate and be of no further force or effect upon any such termination (except that, in the case of termination on a country-by-country basis, any Sublicense granted by Mundipharma to a Third Party in any country in which
(iii)
Mundipharma has not terminated this Agreement shall continue in full force and effect in accordance with the terms of this Agreement and the applicable Sublicense agreement);
(iv) If Mundipharma or its Affiliate had generated any Expanded Licensed Product Clinical Efficacy Data with respect to any Expanded Licensed Product in respect of which the parties had not executed an Expanded Licensed Product Amendment prior to such termination, then, if Cidara wishes to use such Expanded Licensed Product Clinical Efficacy Data in support of any NDA, MAA or other Product Filing or Regulatory Approval for the applicable Expanded Licensed Product or in the commercialization of such Expanded Licensed Product anywhere in the world, Cidara must pay a buy-in fee equal to […***…]. Promptly upon written request by Cidara, Mundipharma shall deliver a written invoice to Cidara for the applicable buy-in fee, and Cidara shall pay the invoiced amount within 30 days of invoice;
(v) Mundipharma shall, and hereby does, effective on such termination, assign to Cidara all of Mundipharma’s and its Affiliates’ right, title and interest in and to the Licensed Product Trademarks (if other than the Cidara Licensed Product Trademark), including, in each case, all goodwill therein, and Mundipharma shall promptly take such actions and execute such instruments, assignments and documents as may be necessary to effect, evidence, register and record such assignment;
(vi) Subject to Cidara’s rights under Section 10.5(d)(vi), Mundipharma may dispose of all inventory of Licensed Product remaining in Mundipharma’s or its Affiliates’ possession as of the effective date of termination for up to six (6) months after the effective date of termination, subject to Mundipharma’s royalty payment, Net Sales Milestone Payment and reporting obligations to Cidara under Section 5.6, Section 5.7 (subject to Sections 5.8 through 0) and Article 6, respectively; and
(vii) Cidara shall have the right, but not the obligation, to purchase from Mundipharma any or all usable inventory of Licensed Product in Mundipharma’s or its Affiliates’ possession as of the date of termination at a supply price equal to […***…]. Any packaging, transport, insurance and other costs relating to delivery shall be at Cidara’s expense.
(e) Termination by Cidara Pursuant to Section 9.4(a), 10.2(a) or 10.2(b). In addition to the effects of termination set forth in Section 10.5(b), in the event of termination of this Agreement by Cidara pursuant to Section 9.4(a), Section 10.2(a) or Section 10.2(b), the following provisions shall apply:
(i) The Grant-Back License shall automatically become worldwide and exclusive both within and outside the Field with respect to Compound and Product;
(ii) Mundipharma shall transfer to Cidara any Expanded Licensed Product Clinical Efficacy Data with respect to any Expanded Licensed Product in respect of which
(iii)
the parties had not executed an Expanded Licensed Product Amendment prior to such termination […***…];
(iv) Mundipharma shall, and hereby does, effective on such termination, assign to Cidara all of Mundipharma’s and its Affiliates’ right, title and interest in and to the Licensed Product Trademarks (if other than the Cidara Licensed Product Trademark), including, in each case, all goodwill therein, and Mundipharma shall promptly take such actions and execute such instruments, assignments and documents as may be necessary to effect, evidence, register and record such assignment;
(v) Subject to Cidara’s rights under Section 10.5(e)(v), Mundipharma may dispose of all inventory of Licensed Product remaining in Mundipharma’s or its Affiliates’ possession as of the effective date of termination for up to six (6) months after the effective date of termination, subject to Mundipharma’s royalty payment, Net Sales Milestone Payment and reporting obligations to Cidara under Section 5.6, Section 5.7 (subject to Sections 5.8 through 0) and Article 6, respectively; and
(vi) Cidara shall have the right, but not the obligation, to purchase from Mundipharma any or all usable inventory of Licensed Product in Mundipharma’s or its Affiliates’ possession as of the date of termination at a supply price equal to […***…]. Any packaging, transport, insurance and other costs relating to delivery shall be at Mundipharma’s expense.
10.6 Manufacturing Technology Transfer. In the event that Cidara requests any manufacturing technology transfer in connection with the exercise of licenses granted to Cidara upon termination of this Agreement in accordance with Section 10.5, […***…].
10.7 Accrued Obligations; Survival. Neither expiration nor termination of this Agreement shall relieve either party of any obligation or liability accruing prior to such expiration or termination, nor shall expiration or termination of this Agreement preclude either party from pursuing all rights and remedies it may have under this Agreement, at law or in equity, with respect to breach of this Agreement. The parties’ rights and obligations under Sections 5.6, 5.7, 5.8, 5.10, 5.11 and 0 (in the case of each of the foregoing sections, as applicable to Mundipharma’s rights and obligations under Section 10.5(c)(iv), 10.5(d)(v) or 10.5(e)(iv), as applicable), 5.9, 7.1, 7.2, 7.3, 7.4, 7.5, 8.1, 9.4(a) (excluding the termination right contained therein), 9.4(b), 9.7, 9.8, 10.3 (if applicable), 10.5, 10.6, 10.7, 13.2(a), 13.3, 13.4, 13.5, 13.6, 13.7, 13.8, 13.9, 13.10 and 13.12, and Articles 1 (to the extent necessary for the interpretation of the other surviving Sections and Articles hereof), 6, 11 and 12, of this Agreement shall survive expiration or termination of this Agreement.
11. | INDEMNIFICATION |
11.1 By Mundipharma. Mundipharma hereby agrees to save, defend and hold Cidara […***…] (each, a “Cidara Indemnitee”) harmless from and against any and all claims, suits, actions, demands, liabilities, expenses and/or loss, including reasonable legal expense and attorneys’ fees (collectively,
11.2
“Losses”) to which any Cidara Indemnitee may become subject as a result of any claim, demand, action or other proceeding by any Third Party to the extent such Losses arise directly or indirectly out of: (a) […***…]; (b) […***…] (c) […***…] or (d) […***…]; except, in each case, to the extent such Losses result from […***…] or […***…].
11.3 By Cidara. Cidara hereby agrees to save, defend and hold Mundipharma […***…] (each, an “Mundipharma Indemnitee”) harmless from and against any and all Losses to which any Mundipharma Indemnitee may become subject as a result of any claim, demand, action or other proceeding by any Third Party to the extent such Losses arise directly or indirectly out of: (a) […***…]; (b) […***…]; (c) […***…]; or (d) […***…]; except, in each case, to the extent such Losses result from […***…] or […***…].
11.4 Procedure. In the event a party (the “Indemnified Party”) seeks indemnification under Section 11.1 or 11.2, the Indemnified Party shall: (a) inform the other party (the “Indemnifying Party”) of a claim as soon as reasonably practicable after it receives notice of the claim (it being understood and agreed, however, that the failure by an Indemnified Party to give notice of a claim as provided in this Section 11.3 shall not relieve the Indemnifying Party of its indemnification obligation under this Agreement except and only to the extent that such Indemnifying Party is actually damaged as a result of such failure to give notice); (b) permit the Indemnifying Party to assume direction and control of the defense of the claim (including the right to settle the claim solely for monetary consideration), using counsel reasonably satisfactory to the Indemnified Party, at the Indemnifying Party’s sole cost and expense; and (c) cooperate as requested (at the expense of the Indemnifying Party) in the defense of the claim. If the Indemnifying Party does not assume control of such defense within […***…] after receiving notice of the claim from the Indemnified Party, the Indemnified Party shall control such defense […***…] The party not controlling such defense may participate therein […***…]. The party controlling such defense shall keep the other party advised of the status of such action, suit, proceeding or claim and the defense thereof and shall consider recommendations made by the other party with respect thereto. The Indemnified Party shall not […***…] . The Indemnifying Party shall not […***…].
11.5 Insurance. Each party, at its own expense, shall maintain product liability and other appropriate insurance (or self-insure) in an amount consistent with Applicable Law and industry standards during the Term and reasonable in light of the activities being conducted by such party under this Agreement. Each party shall provide a certificate of insurance (or evidence of self-insurance) evidencing such coverage to the other party upon request.
12. | DISPUTE RESOLUTION |
12.1 Disputes. Subject to Sections 3.1(f)(v), 12.4 and 12.5, upon the written request of either party to the other party, any claim, dispute, or controversy as to the breach, enforcement, interpretation or validity of this Agreement will be referred to the Senior Executive of Cidara and the Senior Executive of Mundipharma, for resolution. In the event the two individuals referred to in the preceding sentence are unable to resolve such matter within 30 days after the initial written request, then, upon the written demand of either party, the matter shall be subject to arbitration, as provided in Section 12.2, except as expressly set forth in Sections 3.1(f)(v), 12.4 and 12.5.
12.2 Arbitration.
(a) Subject to Sections 3.1(f)(v), 12.4 and 12.5, any dispute, controversy or claim that is not resolved pursuant to Section 12.1 shall be resolved exclusively by final and binding arbitration in accordance with the applicable rules of the International Chamber of Commerce (“ICC”) as then in effect (the “Rules”), except to the extent any such Rule conflicts with the express provisions of this Article 12.
(b) The arbitration shall be conducted by an arbitral tribunal of three neutral arbitrators selected by the ICC in accordance with the Rules; provided that: (i) such arbitrators shall not be current or former employees or directors, or current stockholders, of either party, any of their respective Affiliates or any Sublicensee; (ii) each arbitrators shall have experience and familiarity with commercial licensing practices in the pharmaceutical and biotechnology industries; and (iii) to the extent permitted by the Rules, each party shall have the right to reject up to three proposed arbitrators selected by the ICC. The place of arbitration shall be New York, New York, USA, and all proceedings and communications shall be in the English language.
(c) The arbitral tribunal shall […***…] as reasonably necessary for an understanding of any legitimate issue raised in the arbitration, while also taking into account the desirability of making discovery efficient and cost-effective.
(d) The arbitral tribunal shall, in rendering an award, apply the substantive law of the State of New York, USA, in accordance with Section 13.3 and without giving effect to any conflicts of law provisions thereof that might otherwise refer construction or interpretation of this Agreement to the substantive law of another jurisdiction, and without giving effect to any of its rules or laws relating to arbitration. The award shall include a written statement describing the essential findings and conclusions upon which the award is based, including the calculation of any damages awarded. The arbitral tribunal’s authority to award special, incidental, consequential or punitive damages shall be subject to the limitation set forth in Section 9.8, except to the extent the substantive laws of the State of New York, USA, do not permit such limitation[…***…]. The award rendered by the arbitral tribunal shall be final, binding and non-appealable (subject only to the parties’ right to request correction of any errors in computation, clerical or typographical errors, or other errors of a similar nature, and the arbitral tribunal’s right to make any such correction on its own initiative, in each case, in accordance with the Rules), and judgment upon the award may be entered in any court of competent jurisdiction.
(e) Each party shall bear its own attorneys’ fees, costs, and disbursements arising out of the arbitration, and shall pay an equal share of the fees and costs of the arbitration.
12.3 Confidentiality of Arbitration. Except to the extent necessary to confirm or enforce an award or as may be required by Applicable Law or the rules of any exchange on which a party’s securities are traded, neither a party nor the arbitral tribunal may disclose the existence, content, or results of an arbitration without the prior written consent of both parties.
12.4 Injunctive Relief; Court Actions. Either party may apply to the arbitrators for interim injunctive relief until the arbitration award is rendered or the controversy is otherwise resolved. Either party also may, without waiving any remedy under this Agreement, seek from any court having jurisdiction any injunctive or other equitable relief in the context of a bona fide emergency or prospective irreparable harm, and such an action may be filed and maintained notwithstanding any ongoing discussions between the parties or any ongoing arbitration proceeding. In addition, either party may bring an action in any court of competent jurisdiction to resolve disputes pertaining to the validity, construction, scope, enforceability, infringement or other violations of Patents or other intellectual property rights, and no such claim shall be subject to arbitration pursuant to Section 12.2. Further, no claim under any antitrust, anti-monopoly or
12.5
competition law or regulation, whether or not statutory, shall be subject to arbitration pursuant to Section 12.2.
12.6 Forecast Dispute Resolution. In the event the Senior Executives are unable to agree upon (a) the Peak Incremental Net Sales Forecasts in accordance with Section 4.3(a)(i), or (b) the Subcutaneous Peak Incremental Net Sales Forecasts, including the methodology to be used to determine the Subcutaneous Peak Incremental Net Sales Forecasts, in accordance with Section 4.4(c)(ii), or (c) the Other Product Peak Incremental Net Sales Forecasts, mutatis mutandis, including the methodology to be used to determine such Other Product Peak Incremental Net Sales Forecasts, in accordance with Section 4.4(c)(ii), then, in each case ((a) through (c)), upon the request of either party, the matter will be referred to a neutral Third Party expert with relevant experience and expertise (the “Independent Expert”) mutually agreed to by the parties in good faith (e.g., IQVIA, L.E.K.); provided that if the parties are unable to reach agreement as to the Independent Expert, then each party shall promptly designate one neutral Third Party expert with such experience and expertise, and such experts shall select the Independent Expert by mutual agreement. Within 10 days following selection of the Independent Expert, each party shall submit to the Independent Expert such party’s good faith proposal as to (i) the amounts of the Peak Incremental Net Sales Forecasts pursuant to (a) through (c) above, as applicable, or (ii) the methodology to be used to determine the Peak Incremental Net Sales Forecasts pursuant to (b) and (c) above, as applicable, and (iii) such party’s written argument, not to exceed five pages in length, in favor of the reasonableness of its proposal. Within 15 days following submission of the parties’ written proposals to the Independent Expert, the Independent Expert shall issue a written determination setting forth the Peak Incremental Net Sales Forecast that it believes is the most reasonable […***…] (the “Expert Forecast”), and the following shall apply:
(A) […***…]
(B) […***…]
(C) […***…]
If one of the parties fails to submit its proposal within […***…] after the selection of the Independent Expert, then, […***…], the Independent Expert must select the latter’s proposal. The proposal selected by the Independent Expert, or as determined in accordance with (A) through (C) above, shall be final and binding upon both parties. The parties shall […***…].
13. | MISCELLANEOUS |
13.1 Standstill. Mundipharma hereby agrees that, during the Term, it will not directly or indirectly (including through any Affiliate) acquire beneficial ownership of any equity securities
13.2
of Cidara or securities convertible into or exchangeable for such equity securities, or rights or options to purchase such securities, or acquire the right to control directly or indirectly voting with respect to any other voting securities of Cidara (in each case, other than any such securities of Cidara directly and originally issued by Cidara to Mundipharma or its Affiliate pursuant to a written agreement between Cidara and Mundipharma or its Affiliate), without the prior written consent of Cidara, if the effect of such acquisition would be to entitle Mundipharma to cast directly or indirectly 20% or more of the voting power in any election of directors of Cidara. For purposes of the 20% calculation under this Section 13.1, all such securities, rights or options beneficially owned by Mundipharma (including through Affiliates or others) shall be treated on an as-exercised and as-converted basis, but such securities, rights or options beneficially owned by others shall not be so treated. Mundipharma will not be deemed to have breached this Section 13.1 by reason of (a) Mundipharma’s acquisition of or merger with a Third Party that already owns such securities, rights or options or (b) an acquisition of or merger with Mundipharma by a Third Party that already owns such securities, rights or options. In such event, however, this Section 13.1 shall still apply to activities by Mundipharma and its Affiliates after such event.
Notwithstanding the foregoing, the restrictions provided above shall no longer apply from the earliest of (x) the date Cidara publicly announces that it is seeking an acquisition or business combination in which Cidara would not be the surviving entity, (y) the date Cidara publicly announces its entrance into an agreement or letter of intent with a Third Party other than Mundipharma (or any of its Affiliates) which provides for the acquisition or business combination with Cidara or any of its Affiliates in which Cidara would not be the surviving entity, or (z) the date a Third Party commences a tender offer for all or some lesser portion of Cidara’s common equity that would result in a change of control in Cidara; provided, however, that, in each case, if such efforts or activities cease without any such proposed transaction having been consummated, then the restrictions set forth in the preceding paragraph of this Section 13.1 shall again be applicable.
13.3 Rights Upon Bankruptcy.
(a) Section 365(n). The parties acknowledge and agree that all rights and licenses granted under or pursuant to this Agreement to Mundipharma or Cidara are, and shall otherwise be deemed to be, for purposes of Section 365(n) of the United States Bankruptcy Code and other similar foreign laws, licenses of rights to “intellectual property” as defined under Section 101 of the United States Bankruptcy Code or other similar foreign laws. The parties agree that the parties shall retain and may fully exercise all of their rights and elections under the United States Bankruptcy Code (or any comparable provision of the laws applicable to bankruptcies or insolvencies), and other similar foreign laws. The parties further agree that, in the event of the commencement of a bankruptcy proceeding by or against a party under the United States Bankruptcy Code, the non‑debtor party shall be entitled to a complete duplicate of (or complete access to, as appropriate) any such intellectual property and all embodiments of such intellectual property and the same, which, if not already in the non‑debtor party’s possession, shall be promptly delivered to it (a) upon any such commencement of a bankruptcy proceeding upon the non‑debtor party’s written request therefor, unless the debtor party continues to perform all of its obligations under this Agreement or (b) if not delivered under clause (a) above, following the rejection of this Agreement by or on behalf of the debtor party upon written request therefor by the non‑debtor party.
(b) […***…]
(i) […***…]
(ii) […***…]
(1) […***…]
(2) […***…]
(iii) […***…]
13.4 Governing Law. This Agreement and any disputes, claims, or actions related thereto shall be governed by and construed and enforced in accordance with the laws of the State of New York, USA, without regard to any conflicts of law provisions thereof that might otherwise refer construction or interpretation of this Agreement to the substantive law of another jurisdiction. The parties agree to exclude the application to this Agreement of the United Nations Convention on Contracts for the International Sale of Goods.
13.5 Entire Agreement; Amendment. This Agreement, including the Exhibits hereto, is both a final expression of the parties’ agreement and a complete and exclusive statement with respect to all of its terms. This Agreement supersedes all prior and contemporaneous agreements and communications, whether oral, written or otherwise, concerning any and all matters contained
13.6
herein. This Agreement may only be modified or supplemented in a writing expressly stated for such purpose and signed by an authorized representative of each party.
13.7 Relationship Between the Parties. The parties’ relationship, as established by this Agreement, is solely that of independent contractors. This Agreement does not create any partnership, joint venture or similar business relationship between the parties. Neither party is a legal representative of the other party, and neither party shall assume or create, or purport to assume or create, any obligation, representation, warranty or guarantee, express or implied, on behalf of the other party for any purpose whatsoever.
13.8 Non-Waiver. The failure of a party to insist upon strict performance of any provision of this Agreement or to exercise any right arising out of this Agreement shall neither impair that provision or right nor constitute a waiver of that provision or right, in whole or in part, in that instance or in any other instance. Any waiver by a party of a particular provision or right shall be in writing, shall be as to a particular matter and, if applicable, for a particular period of time and shall be signed by an authorized representative of such party.
13.9 Assignment. Except as expressly provided hereunder, neither this Agreement nor any rights or obligations hereunder may be assigned or otherwise transferred by either party without the prior written consent of the other party (which consent shall not be unreasonably withheld conditioned or delayed); provided, however, that either party may assign this Agreement and its rights and obligations hereunder without the other party’s consent:
(a) in connection with the transfer or sale of all or substantially all of such party’s business related to Compound and Product to a Third Party […***…], whether by merger, sale of stock, sale of assets or otherwise, provided that in the event of a such transaction (whether this Agreement is actually assigned or is assumed by the acquiring party by operation of law (e.g., in the context of a reverse triangular merger)), provided that:
(i) the acquiring Third Party provides written confirmation to the other party of such Third Party’s agreement to be bound by and to comply with the terms of this Agreement;
(ii) intellectual property rights of the acquiring party to such transaction (if other than one of the parties to this Agreement) shall not be included in the technology licensed hereunder or otherwise subject to this Agreement; and
(iii) notwithstanding any other provision of this Agreement to the contrary, Section 4.6 shall not apply to any Anti-Fungal Product that is: […***…]; provided, however, that, in each case ((A), (B) and (C)), […***…]. For clarity, however, in the event of such acquisition, Section 4.6 shall continue to apply to any Anti-Fungal Product that was […***…]; or
(b) to an Affiliate of such party, provided that the assigning party shall remain liable and responsible to the non‑assigning party hereto for the performance and observance of all such duties and obligations by such Affiliate.
For purposes of Section 13.7(a), […***…].
The rights and obligations of the parties under this Agreement shall be binding upon and inure to the benefit of the successors and permitted assigns of the parties, and the name of a party appearing herein shall be deemed to include the name of such party’s successors and permitted assigns to the extent necessary to carry out the intent of this section. Any assignment not in accordance with this Agreement shall be void.
13.10 No Third Party Beneficiaries. This Agreement is neither expressly nor impliedly made for the benefit of any party other than those executing it. Without prejudice to the foregoing, the parties agree […***…]; provided that the limitations of liability set forth in this Agreement shall […***…].
13.11 Severability. If, for any reason, any part of this Agreement is adjudicated invalid, unenforceable or illegal by a court of competent jurisdiction, such adjudication shall not affect or impair, in whole or in part, the validity, enforceability or legality of any remaining portions of this Agreement. All remaining portions shall remain in full force and effect as if the original Agreement had been executed without the invalidated, unenforceable or illegal part.
13.12 Notices. Any notice to be given under this Agreement must be in writing and delivered either in person, by any method of mail (postage prepaid) requiring return receipt, or by overnight courier or facsimile confirmed thereafter by any of the foregoing, to the party to be notified at its address(es) given below, or at any address such party has previously designated by
13.13
prior written notice to the other. Notice shall be deemed sufficiently given for all purposes upon the earliest of: (a) the date of actual receipt; or (b) if delivered by overnight courier, the next Business Day the overnight courier regularly makes deliveries.
If to Mundipharma: | Mundipharma Medical Company Par La Ville Place 00 Xxx Xx Xxxxx Xxxx Xxxxxxxx XX00 Xxxxxxx Xxxxxxxxx: […***…], General Manager Telephone: […***…] |
With a copy to: | Mundipharma International Limited Unit 196 Cambridge Science Xxxx Xxxxxx Xxxx Xxxxxxxxx XX0 0XX Xxxxxx Xxxxxxx Attention: General Counsel Telephone: […***…] |
If to Cidara: | 0000 Xxxxx Xxxxx Xxxxx, Xxxxx 000 Xxx Xxxxx, XX 00000 XXX Attention: Chief Executive Officer Telephone: […***…] |
With a copy to: | 0000 Xxxxx Xxxxx Xxxxx, Xxxxx 000 Xxx Xxxxx, XX 00000 XXX Attention: General Counsel Telephone: […***…] |
13.14 Force Majeure. Each party shall be excused from liability for the failure or delay in performance of any obligation under this Agreement if such failure is caused by reason of any event beyond such party’s reasonable control including but not limited to acts of nature, fire, flood, explosion, earthquake, or other natural forces, war, civil unrest, acts of terrorism, accident, destruction or other casualty, any lack or failure of transportation facilities, any lack or failure of supply of raw materials, or any other event similar to those enumerated above. Such excuse from liability shall be effective only to the extent and duration of the event(s) causing the failure or delay in performance and provided that the party has not caused such event(s) to occur. Notice of a party’s failure or delay in performance due to force majeure must be given to the other party within 30 days after its occurrence. All delivery dates under this Agreement that have been affected by force majeure shall be tolled for the duration of such force majeure.
13.15 Interpretation. The headings of clauses contained in this Agreement preceding the text of the sections, subsections and paragraphs hereof are inserted solely for convenience and ease of reference only and shall not constitute any part of this Agreement, or have any effect on its interpretation or construction. All references in this Agreement to the singular shall include the plural where applicable. The word “including” (and variations thereof) as used in this Agreement means including, without limiting the generality of any description preceding such term, and the word “or” has the inclusive meaning represented by the phrase “and/or.” Unless otherwise specified, references in this Agreement to any Article shall include all Sections, subsections and paragraphs in such Article, references to any Section shall include all subsections and paragraphs in such Section, and references in this Agreement to any subsection shall include all paragraphs in such subsection. All references to days in this Agreement shall mean calendar days, unless otherwise specified. Ambiguities and uncertainties in this Agreement, if any, shall not be interpreted against either party, irrespective of which party may be deemed to have caused the ambiguity or uncertainty to exist. This Agreement has been prepared in the English language and the English language shall control its interpretation. In addition, all notices required or permitted to be given hereunder, and all written, electronic, oral or other communications between the parties regarding this Agreement shall be in the English language.
13.16 Counterparts. This Agreement may be executed in counterparts, including by transmission of facsimile or PDF copies of signature pages to the parties or their representative legal counsel, each of which shall be deemed an original document, and all of which, together with this writing, shall be deemed one instrument.
[Remainder of this page intentionally left blank.]
IN WITNESS WHEREOF, the parties hereto have duly executed this Collaboration and License Agreement as of the Effective Date.
Mundipharma Medical Company | |
By: Name: Title: | By: Name: Title: |
Exhibit A
Cidara Patents
[…***…]
Exhibit B
Global Development Plan
[…***…]
Exhibit C
JSC Dispute Resolution Matrix re Key Matters
[…***…]
16.
Exhibit D
CMC Development Plan
[…***…]
Exhibit E
Lead Compound
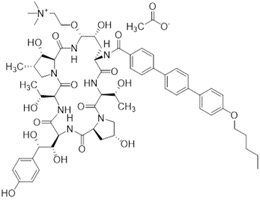
Exhibit F
Mundipharma Territory Plan
[…***…]
Exhibit G
Key Supply Terms
[…***…]
Exhibit H
Form of Press Release


Cidara Therapeutics and Mundipharma Form Strategic Partnership to Develop and Commercialize Rezafungin
Collaboration combines strengths to develop and commercialize life-saving antifungal treatment and prophylaxis, an area of high unmet medical need
Mundipharma acquires exclusive rights to develop and commercialize rezafungin in all markets outside of the United States and Japan, which will be retained by Cidara
Cidara to receive upfront payment of $30 million and equity investment of $9 million, co-development funding, development milestones and tiered royalty stream
Total transaction value could exceed $568 million
Cidara to host conference call today at 8:00 a.m. ET/5:00 a.m. PT
SAN DIEGO, CA AND CAMBRIDGE, UK, September 3, 2019 – Cidara Therapeutics, Inc. (Nasdaq: CDTX) and Mundipharma announced today that they have entered into a strategic partnership to develop and commercialize rezafungin for the treatment and prevention of invasive fungal infections. Rezafungin is a novel, once-weekly echinocandin antifungal being developed for the first-line treatment of candidemia and invasive candidiasis as well as for the prophylaxis of invasive fungal infections in patients undergoing allogeneic blood and marrow transplantation, for which no new therapies have been approved in over 13 years.
The partnership agreement follows Cidara’s recent announcement of the successful completion of its STRIVE B Phase 2 trial. Under the terms of the agreement, in exchange for granting Mundipharma exclusive commercialization rights to rezafungin outside the U.S. and Japan, Cidara will receive a $30 million upfront payment and Mundipharma will make a $9 million equity investment in Cidara. Cidara will also receive an additional $42 million in near-term funding to support the global Phase 3 ReSTORE and ReSPECT trials for the treatment and prevention of fungal infections. In addition, Cidara is eligible to receive development, regulatory
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
and commercial milestone payments, representing a total potential transaction value of $568 million plus double-digit royalties. Cidara will continue to lead the ongoing global Phase 3 development programs for rezafungin with the support of Mundipharma. The companies may pursue additional indications or formulations of rezafungin.
“This is a transformational collaboration for Cidara, and we look forward to working closely with our colleagues at Mundipharma, a highly successful, profitable company with a commercial presence spanning 120 markets worldwide and annual sales exceeding €2 billion,” said Xxxxxxx Xxxxx, Ph.D., President and Chief Executive officer of Cidara. “Mundipharma is particularly well positioned globally with established hospital and hematology/oncology business units to fully leverage the commercial potential of rezafungin. Through this partnership, both companies fully commit to advancing rezafungin and helping to save the lives of patients who are highly vulnerable to these deadly infections.”
“By partnering with Cidara on rezafungin, we continue to serve our purpose - to move medicine forward,” said Xxxxxxx Xxxxxxxx, Ph.D., M.B.A., President and Chief Executive officer of Mundipharma. “In a world where antifungal resistance is posing a major threat to the lives of vulnerable immunocompromised patients, rezafungin shows promise to address a major unmet medical need as well as potentially providing a wider spectrum of efficacy in a more convenient administration schedule. With our proven commercial excellence we are confident that we will maximize the potential of this differentiated and innovative asset. Rezafungin will be a significant addition to our pipeline that integrates well with our overall portfolio and sales force capabilities. We are excited to work with the team at Cidara to deliver such an important medicine to patients around the world.”
Conference Call and Webcast
Cidara management will host a conference call and webcast at 8:00 a.m. ET/5:00 a.m. PT today. The live call may be accessed by dialing (000) 000-0000 for domestic callers and (000) 000-0000 for international callers and entering the conference code: 6567991. The webcast will be made available on Cidara’s website at xxx.xxxxxx.xxx under the Investors tab in the Events section. Following the live audio webcast, a replay will be available on Cidara's website.
About Invasive Fungal Infections
Invasive fungal infections (IFIs) represent a serious threat to millions of patients worldwide, resulting in more than 1.5 million deaths annually and mortality rates ranging from 15 to 65 percent. These infections continue to be a global health issue, especially for critically ill patients in hospitals and patients with compromised immune systems, including cancer and transplant patients. Approximately 90 percent of IFI-related deaths are associated with Candida, Aspergillus, and Pneumocystis.
H-2
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
About Rezafungin
Rezafungin is a novel echinocandin antifungal and the only once-weekly drug candidate being developed for the first-line treatment and prevention of serious invasive fungal infections. Rezafungin has a unique pharmacokinetic profile with a prolonged half-life and front-loaded plasma exposure which, in contrast to all other echinocandins, allows for once-weekly IV therapy for inpatient and outpatient use. The U.S. Food and Drug Administration (FDA) has designated rezafungin as a Qualified Infectious Disease Product (QIDP) with Fast Track status and orphan drug designation related to its use in the treatment of candidemia and invasive candidiasis.
About Cidara Therapeutics
Cidara is a clinical-stage biotechnology company focused on the discovery, development and commercialization of novel anti-infectives that have the potential to transform the standard of care and save or improve patients’ lives. Cidara is currently advancing its novel echinocandin antifungal, rezafungin acetate, in a Phase 3 clinical trial for the first-line treatment of candidemia and/or invasive candidiasis (ReSTORE) and plans to commence a second Phase 3 trial of once-weekly rezafungin for prophylaxis against invasive fungal infections in patients undergoing allogeneic blood and marrow transplantation (ReSPECT) initially in Europe and Canada. In addition to its robust rezafungin clinical program, Cidara is applying its proprietary Cloudbreak® platform to develop antiviral conjugates (AVCs) for the prevention and treatment of influenza and other viral diseases. The Cloudbreak platform is designed to discover compounds that both directly kill pathogens and direct a patient’s immune system to attack and eliminate pathogens. Cidara is headquartered in San Diego, California. For more information, please visit xxx.xxxxxx.xxx.
About Mundipharma
Mundipharma is a global network of privately-owned independent associated companies whose purpose is to move medicine forward.
With a high performing and learning organisation that strives for innovation and commercial excellence through partnerships, we successfully transformed and diversified our portfolio of medicines to create value for patients, payers and wider healthcare systems across important therapeutic areas such as Diabetes, Respiratory, Oncology, Pain and Biosimilars.
For more information please visit: xxx.xxxxxxxxxxx.xxx
Forward-Looking Statements
Statements contained in this press release regarding matters that are not historical facts are "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act
H-3
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
of 1995. Because such statements are subject to risks and uncertainties, actual results may differ materially from those expressed or implied by such forward-looking statements. Such statements include, but are not limited to, statements relating to the transformational nature and value of Cidara’s collaboration with Mundipharma, Cidara’s ability to develop new anti-infectives that are innovative or address unmet needs, including Cidara’s ability to successfully complete the ReSTORE and ReSPECT Phase 3 clinical trials, Cidara’s ability to complete development of, obtain regulatory approval for and commercialize rezafungin including Cidara’s ability to receive milestone payments for the achievement of development milestones, the potential for rezafungin to successfully treat or prevent invasive fungal infections and represent an improvement over current approaches, and the ability of Cidara’s Cloudbreak program to successfully identify and develop product candidates to prevent and/or treat viral diseases, and other diseases. Risks that contribute to the uncertain nature of the forward-looking statements include: the success and timing of Cidara’s clinical trials; regulatory developments in the United States and foreign countries; changes in Cidara’s plans to develop and commercialize its product candidates; Cidara’s ability to obtain additional financing; Cidara’s ability to obtain and maintain intellectual property protection for its product candidates; and the loss of key scientific or management personnel. These and other risks and uncertainties are described more fully in Cidara’s most recent filings with the United States Securities and Exchange Commission. All forward-looking statements contained in this press release speak only as of the date on which they were made. Cidara undertakes no obligation to update such statements to reflect events that occur or circumstances that exist after the date on which they were made.
CIDARA - INVESTOR CONTACT:
Xxxxxx X. Xxx
Westwicke Partners, LLC
Managing Director
(000) 000-0000
xxxxxx.xxx@xxxxxxxxx.xxx
CIDARA - MEDIA CONTACT:
Xxxxxx Xxxxx
Xxx Xxxxx Inc.
(000) 000-0000
xxxxxxxxxxx@xxxxxxxx.xxx
MUNDIPHARMA - MEDIA CONTACTS:
Xxxxx Xxxxxxxx
Xxxxxx Health Communications Limited
x00 (0) 00 00 000 000
xxxxx@xxxxxxxxxxxx.xxx
H-4
CERTAIN CONFIDENTIAL PORTIONS HAVE BEEN REDACTED FROM THIS EXHIBIT BECAUSE THEY ARE BOTH (i) NOT MATERIAL AND (ii) WOULD BE COMPETITIVELY HARMFUL IF PUBLICLY DISCLOSED. INFORMATION THAT HAS BEEN OMITTED HAS BEEN IDENTIFIED IN THIS DOCUMENT WITH A PLACEHOLDER IDENTIFIED BY THE XXXX “[***]”.
Xxxxxxx Grand
Mundipharma
x00 (0) 0000 000 000
xxxxxxx.xxxxx@xxxxxxxxxxx.xxx
###
H-5

