
MASTER SERVICES AGREEMENT Between Medpace Inc. an Ohio Corporation Cincinnati, Ohio 45212 (“MEDPACE”) and VIVUS, Inc. a Delaware Corporation Mountain View, California 94040-2552 (“VIVUS”)
Exhibit 10.2
Between
Medpace Inc.
an Ohio Corporation
▇▇▇▇ ▇▇▇▇▇▇ ▇▇▇▇▇▇
▇▇▇▇▇▇▇▇▇▇, ▇▇▇▇ ▇▇▇▇▇
(“MEDPACE”)
and
VIVUS, Inc.
a Delaware Corporation
▇▇▇▇ ▇▇▇▇▇▇ ▇▇▇▇▇▇
▇▇▇▇▇▇▇▇ ▇▇▇▇, ▇▇▇▇▇▇▇▇▇▇ ▇▇▇▇▇-▇▇▇▇
(“VIVUS”)
This MASTER SERVICES AGREEMENT (the “Agreement”), dated as of September 12, 2007 (the “Effective Date”), is between MEDPACE and VIVUS. MEDPACE and VIVUS are sometimes referred to herein individually as a “Party” and together as the “Parties”.
RECITALS:
WHEREAS, VIVUS is in the business of developing and obtaining regulatory approval of the marketing and sale of pharmaceutical products and
WHEREAS, MEDPACE is engaged in the business of providing services related to the design and execution of clinical development programs involving drugs, biologics, and medical devices through engagement by its clients, the sponsors of clinical development programs, to perform such services; and
WHEREAS, VIVUS desires to engage MEDPACE to perform certain services (“Services”) as set forth hereinafter in connection with certain clinical trials, all in accordance with and subject to the terms of this Agreement;
NOW, THEREFORE, in consideration of the premises and the mutual covenants and conditions hereinafter set forth, the Parties agree as follows:
1. PROJECT SPECIFICATIONS
1.1 MEDPACE hereby agrees to perform Services for VIVUS from time to time. The precise Services to be performed by MEDPACE shall be mutually agreed upon by the Parties and set forth in one or more task orders (each a “Task Order”), a form of which is attached hereto as Exhibit A. Each Task Order shall be signed by an authorized representative of each Party and shall include detailed information concerning a given project, including a description of the specific services to be provided (“Scope of Work”), project milestones and target completion dates (“Project Schedule”), a detailed budget (“Project Budget”), and a schedule of payments related to the Project Schedule and the Project Budget (“Payment Schedule”). Each Task Order shall contain a Transfer of Obligations list (“Transfer of Obligations”) in conjunction with the relevant Task Order and consistent with the regulations set forth in 21 C.F.R. Section 312, Subpart D (Responsibilities of VIVUS and Investigators). Any responsibilities not specifically transferred in the Transfer of Obligations shall remain the regulatory responsibility of VIVUS. Each Task Order shall designate a Project Manager or other duly authorized representative authorized to make decisions on behalf of each Party with respect to the Services to be rendered under the Task Order.
2. PROJECT SCHEDULE
2.1. Each Task Order shall contain project timelines, milestones or target dates for completion of a project or a portion thereof, and all such schedules shall be reasonable for the Services to be provided. In all events, the Parties shall use their reasonable best efforts to comply with each Task Order.
2.2. If at any time either Party anticipates a delay in meeting the timelines for a given Task Order as set forth in its Project Schedule, either due to changes to the Services requested by VIVUS, or other causes (such as FDA approval of a competitor’s NDA for the same drug, which may adversely affect patient enrollment), then the anticipating Party shall promptly notify the other Party in writing, specifying the reason for the delay and the anticipated effect upon the timelines, milestones or other deliverables.
3. CHANGE ORDERS
3.1. Any change in the details of a Task Order or the assumptions upon which the Task Order is based may require changes in the Project Budget, Payment Schedule or Project Schedule. Every such change shall require a written amendment to the Task Order (a “Change Order”). Each Change Order shall detail the requested changes to the applicable task, responsibility, duty, budget, timeline or other matter. The Change Order will become effective upon the execution of the Change Order by both Parties, and the Change Order will specify the period of time within which MEDPACE must implement the changes. Both Parties agree to act in good faith and promptly when considering a Change Order requested by the other party but neither party is obligated to execute a Change Order. No Change Order shall become effective unless and until it is signed by both Parties. Any such changes that result in additional charges shall be reflected in the Change Order to the affected Task Order, Project Budget or Payment Schedule.
4. PROJECT BUDGET, PAYMENT SCHEDULE, AND TERMS
4.1. VIVUS agrees to pay MEDPACE for Services rendered pursuant to the Project Budget and Payment Schedules included in each Task Order.
4.2. VIVUS agrees to reimburse MEDPACE for reasonable pass-through expenses identified in the Task Order and incurred by MEDPACE in providing the Services in accordance with the relevant Task Order. All expenses billed to VIVUS by MEDPACE must be accompanied by appropriate documentary evidence, such as receipts or other documentation reasonably acceptable to VIVUS.
4.3. VIVUS shall mail payments to MEDPACE within 45 days after receipt of a written invoice and required supporting documentation as applicable. An annual interest rate of 12% will be applied to outstanding invoices greater than 45 days.
5. WARRANTIES AND REPRESENTATIONS:
5.1. Acknowledgements:
MEDPACE acknowledges that the Services to be provided hereunder are for the benefit of, and are subject to the direction of VIVUS. MEDPACE acknowledges that VIVUS is the beneficiary under the terms of this Agreement and each Task Order, and that VIVUS is entitled to enforce the provisions thereof.
5.2. Representations and Warranties of MEDPACE
5.2.1. MEDPACE represents and warrants that it is a corporation with its principal office and place of business at ▇▇▇▇ ▇▇▇▇▇▇ ▇▇▇▇▇▇, ▇▇▇▇▇▇▇▇▇▇, ▇▇▇▇ ▇▇▇▇▇, duly organized, validly existing and in good standing in its place of organization, and is in good standing in and duly qualified to do business.
5.2.2. MEDPACE warrants that the execution, delivery and performance of this Agreement and each Task Order has been validly authorized by all corporate action and this Agreement and each Task Order represents the valid binding agreement of MEDPACE enforceable in accordance with its terms. The execution, delivery and performance of this Agreement and each Task Order will not violate any organizational document governing MEDPACE, any agreement to which MEDPACE is a party, or any law or court or governmental order, holding or writ by which MEDPACE is bound. MEDPACE further warrants that it shall render the Services requested by VIVUS in accordance with high professional standards, consistent with Good Clinical Practices and Laboratory Regulations and with the standard of care customary in the contract research organization industry. MEDPACE shall complete all services in conformance with each approved Task Order, including each Project Schedule and Project Budget.
5.2.3. MEDPACE warrants that the personnel assigned to perform services rendered under this Agreement shall be qualified and professionally capable of performing the Services, shall be adequate to effectively perform the Services on the agreed upon schedule and shall devote such time as is necessary to perform the Services on such agreed upon schedule.
5.2.4. MEDPACE further warrants that it shall perform the Services in compliance with the terms of this Agreement, the terms of the Task Orders, and all applicable laws and regulations including, without limitation, the Federal Food, Drug and Cosmetic Act and the regulations promulgated pursuant thereto, and all future amendments during the term. MEDPACE further warrants that it shall make available to VIVUS, or to the responsible regulatory authority, relevant records, programs and data as may reasonably be requested by VIVUS or which is the subject of a Task Order. VIVUS shall have the right to monitor the operations of MEDPACE hereunder, and VIVUS representatives shall have the right to visit any of the facilities where MEDPACE is performing any of the Services and during such visits to inspect the work being done and materials used, to observe the procedures being followed, to examine the books, records and other data relevant to the Services. If any regulatory agency requests to inspect any books, records, data or facilities of
MEDPACE relating to the Services, MEDPACE shall immediately notify VIVUS.
5.2.5. MEDPACE represents and warrants that there is no litigation, regulatory investigation or proceeding, administrative hearing or any other similar proceeding pending or to the best of its knowledge threatened against MEDPACE which could adversely affect MEDPACE’s ability to perform the Services or which alleges a violation by MEDPACE of any law or regulation related to Services.
5.2.6. Obligation of MEDPACE
MEDPACE shall immediately notify VIVUS in writing if any of the representations or warranties contained in this Article 5 become untrue. MEDPACE shall correct or perform again any portion of the Services that fails to conform to any warranty set forth in this Article 5 without additional cost to VIVUS and within thirty (30) days of receipt of notice from VIVUS.
5.3. Representations and Warranties of VIVUS
5.3.1 VIVUS represents and warrants that it is a corporation with its principal office and place of business at ▇▇▇▇ ▇▇▇▇▇▇ ▇▇▇▇▇▇, ▇▇▇▇▇▇▇▇ ▇▇▇▇, ▇▇▇▇▇▇▇▇▇▇ ▇▇▇▇▇-▇▇▇▇, duly organized, validly existing and in good standing in its place of organization, and is in good standing in and duly qualified to do business.
5.3.2 VIVUS warrants that the execution, delivery and performance of this Agreement and each Task Order has been validly authorized by all corporate action and this Agreement and each Task Order represents the valid binding agreement of VIVUS enforceable in accordance with its terms. The execution, delivery and performance of this Agreement and each Task Order will not violate any organizational document governing VIVUS, any agreement to which VIVUS is a party, or any law or court or governmental order, holding or writ by which VIVUS is bound.
5.3.3 VIVUS represents and warrants that there is no litigation, regulatory investigation or proceeding, administrative hearing or any other similar proceeding pending or to the best of its knowledge threatened against VIVUS which could adversely affect VIVUS’s ability to perform under this Agreement or any Task Order or which alleges a violation by VIVUS of any law or regulation related to the subject matter of the Services rendered by MEDPACE hereunder.
5.3.4 Obligation of VIVUS
VIVUS shall immediately notify MEDPACE in writing if any of the representations or warranties contained in this Article 5 become untrue.
6. TERMINATION
6.1 VIVUS may terminate this Agreement without cause immediately upon giving MEDPACE notice of such termination, provided such termination shall not in and of itself affect any then uncompleted Task Order.
6.2 VIVUS may terminate any Task Order without cause immediately upon giving MEDPACE notice of such termination. As soon as practicable, after receipt of such notice, the Parties shall cooperate in good faith to agree on a plan to expeditiously conclude activities with respect to such matter. MEDPACE shall transfer to VIVUS all case report forms, study files, and other data and information in any and all formats available, including electronic format and computer files and programs, in MEDPACE’s possession.
6.3 MEDPACE may terminate a Task Order only if VIVUS has defaulted on its obligations thereunder and has not cured such default within 10 days after written notice if the default is the failure to pay MEDPACE any amount due thereunder or within 30 days after written notice in the event of any other default, upon giving VIVUS notice of such termination; provided, however, that such amounts are not in dispute. Should VIVUS dispute any amounts due, the undisputed portion will be paid in accordance with Section 4.5, and VIVUS and MEDPACE will work together in good faith to settle the disputed portion. Upon resolution, any amounts then owing will be paid within ten (10) days of reaching said resolution. As soon as practicable, after receipt of such notice, the Parties shall cooperate in good faith to agree on a plan to expeditiously conclude activities with respect to such matter.
MEDPACE shall transfer to VIVUS all case report forms, study files, and other data and information in any and all formats available, including electronic format and computer files and programs, in MEDPACE’s possession.
6.4 In the event of any termination of a Task Order before completion, VIVUS agrees to pay MEDPACE for all Services rendered pursuant to the unfinished Task Order prior to such termination and any non-cancelable expenses incurred in connection with MEDPACE’s performance of Services thereunder. As soon as reasonably practicable following receipt of a termination notice, MEDPACE shall submit an itemized accounting of Services performed, expenses incurred pursuant to performance of the Services, non-cancelable expenses incurred by MEDPACE relating to any unfinished Task Order, and payments received in order to determine a balance to be paid by either Party to the other. Such balance shall be paid within 30 days of receipt of such an itemized accounting by VIVUS.
7. COMMUNICATIONS
7.1 Any notice required or permitted under this Agreement shall be in writing and shall be deemed given if delivered personally, mailed by prepaid, first class, certified mail, return receipt requested, or sent by express courier service, to the
Party to be notified at the addresses set forth below (or such other address as shall be designated by written notice); provided that all notices shall be effective upon receipt thereof:
If to MEDPACE:
Medpace, Inc.
▇▇▇▇ ▇▇▇▇▇▇ ▇▇▇▇▇▇
▇▇▇▇▇▇▇▇▇▇, ▇▇▇▇ ▇▇▇▇▇
Attn: August ▇. ▇▇▇▇▇▇▇▇
Telephone: (▇▇▇) ▇▇▇-▇▇▇▇ ▇▇▇▇▇
If to VIVUS:
VIVUS, Inc.
▇▇▇▇ ▇▇▇▇▇▇ ▇▇▇▇▇▇
▇▇▇▇▇▇▇▇ ▇▇▇▇, ▇▇ ▇▇▇▇▇
Attn: Legal Affairs
Telephone: (▇▇▇) ▇▇▇-▇▇▇▇
8. CONFIDENTIALITY
8.1. VIVUS may provide confidential information to MEDPACE during the course of this Agreement. All information by VIVUS or its clients or data collected by MEDPACE for VIVUS during the course of performance of the Services is deemed to be the confidential information of VIVUS. MEDPACE shall not disclose confidential information to any third party, or use the confidential information for any purpose other than for the benefit of VIVUS, without the prior written consent of VIVUS.
8.1.1. MEDPACE shall ensure by binding written agreement that its employees, agents, and approved independent contractors involved in the Services shall comply with the provisions of Article 8 of this Agreement. MEDPACE shall disclose only confidential information to those of its employees, agents, and independent contractors who reasonably need to know the confidential information.
8.1.2. MEDPACE shall exercise due care, but no less than a reasonable degree of care, to prevent the unauthorized disclosure and use of confidential information associated with the Services.
8.2. MEDPACE Confidential Information.
VIVUS shall hold confidential all non-public information and materials furnished to it by MEDPACE pertaining to MEDPACE’s business practices or pertaining to proprietary information of MEDPACE.
8.3. This confidentiality and nondisclosure provision shall not apply to:
Information which was known by the Party before the date hereof or which is independently discovered, after the date hereof, without the aid, application or use of the confidential information, as evidenced by written records;
Information which is in the public domain on the date hereof or subsequently becomes publicly available through no fault or action of the other Party; or
Information, which is disclosed to the Party by a third party authorized to disclose it.
8.3.1. If the receiving Party is requested to disclose the Confidential Information of the other Party or the substance of this Agreement in connection with a legal or administrative proceeding or otherwise to comply with a requirement under the law, the receiving party will give the disclosing Party prompt notice of such request so that the disclosing Party may seek an appropriate protective order or other remedy, or waive compliance with the relevant provisions of this Agreement. The disclosing Party must notify the receiving Party within 10 days that it intends to take action in response to the request for disclosure. If the disclosing Party seeks a protective order or other remedy, the receiving Party, at the disclosing Party’s expense, will cooperate with and assist the disclosing Party in such efforts. Failure of the disclosing Party to intervene shall not relieve the obligations to maintain confidentiality except in so far as the receiving Party must comply with the terms of such process compelling disclosure.
9. RIGHTS IN PROPERTY
9.1. All materials, documents, data, software and information of every kind and description supplied to MEDPACE by VIVUS or any of VIVUS’s clients, or prepared, developed, or generated by MEDPACE pursuant to this Agreement, (except for the pre-existing MEDPACE procedural manuals, personal data, methods, procedures, and policies) are and shall be the sole and exclusive property of VIVUS. Further, all data and information generated or derived by MEDPACE as the result of services performed by it under this Agreement shall be and remain the exclusive property of VIVUS. VIVUS shall have the right to make whatever use they deem desirable of any such materials, documents, data or software. MEDPACE shall not, without the prior written consent of VIVUS, publish, disseminate, or otherwise disclose to any third party any such property (except such disclosure as may be required by law), or use any such property for any purpose other than the performance of this Agreement. Any inventions or other intellectual property, including without limitation protectable copyrights and trademarks, that may evolve from the data and information described above or as the result of Services performed by MEDPACE under this Agreement shall belong to VIVUS and MEDPACE agrees to assign its rights in all such inventions and/or other intellectual property to VIVUS consistent with the obligations set forth in Article 10 below.
9.2. VIVUS acknowledges that all computer programs, software, applications, databases, proposals and other documentation generally used by MEDPACE and not directly related to, derived from or developed solely for VIVUS are the exclusive and confidential property of MEDPACE or the third parties from whom MEDPACE has secured the right of use. VIVUS agrees that any improvement, alteration or enhancement to MEDPACE systems, software, applications or processes which are developed or implemented during the course of any Services performed hereunder, without the use of any VIVUS data, information, materials or Confidential Information (or derivatives thereof), shall be the property of MEDPACE.
10. PATENT RIGHTS
10.1. MEDPACE shall disclose promptly to VIVUS any and all inventions, discoveries and improvements conceived or made by MEDPACE while providing such services to VIVUS pursuant to the Agreement and constituting a modification or extension of use relating to VIVUS’s proprietary rights, and agrees to assign all its interest therein to VIVUS or its nominee; whenever requested to do so by VIVUS, MEDPACE shall execute any and all applications, assignments, or other instruments and give testimony which VIVUS shall deem necessary to apply for and obtain a patent in the United States of America and/or other applicable jurisdiction or of any foreign country or to protect otherwise VIVUS’s interests and shall compensate MEDPACE for the time devoted to said activities and reimburse it for expenses incurred.
11. PUBLICITY
11.1. MEDPACE shall not make any public announcements concerning this Agreement or the subject matter hereof without the prior written consent of VIVUS.
11.2 VIVUS may not use MEDPACE’s name, logo or trademark in any communication, release, notice or other publication without the express prior written consent of MEDPACE, except as required by SEC.
12. SECURITY AND DISPOSITION OF STUDY FILES
12.1. MEDPACE shall use commercially reasonable efforts, including, but not limited to, periodic backup of computer files, to prevent the loss or alteration of VIVUS’s study data, Confidential Information, documentation, and correspondence. MEDPACE shall in all respects comply with any Food and Drug Administration regulations concerning the maintenance, creation and storage of records, including electronic records.
12.2. At appropriate time points or at completion of Services under a Task Order, MEDPACE shall transfer study materials, documents and correspondence to VIVUS. MEDPACE shall have the right to retain one copy of any study materials, documentation, and correspondence necessary solely to meet regulatory
or MEDPACE’s own internal audit requirements, so long as it continues to maintain the Confidentiality requirements of Article 8.
13. VIVUS OBLIGATIONS
13.1. VIVUS acknowledges that performance of the Services by MEDPACE will require the co-operative involvement of both Parties, and VIVUS hereby agrees to provide such assistance as may be reasonably necessary to enable MEDPACE to perform the Services.
14. INDEMNIFICATION
14.1 Indemnification by VIVUS
VIVUS agrees to indemnify, defend and hold harmless MEDPACE, and each officer, agent, employee and contractor of Medpace (Medpace and each such party, a “MEDPACE Indemnitee”) from any and all liability, loss, expenses (including reasonable attorneys’ fees) or damage such MEDPACE Indemnitee may suffer (“Damages”) as the result of any claim, demand, cost or judgment against them arising out of the Services to be performed pursuant to each Task Order which arise, or are alleged to arise, except as follows:
(i) A Medpace Indemnitee shall not be entitled to indemnification hereunder to the extent such Damages are the result of the negligence or willful misconduct of such MEDPACE Indemnitee (provided all Medpace Indemnitees who are not negligent or guilty of willful misconduct causing such Damages shall still be entitled to the indemnification hereunder notwithstanding the negligence or willful misconduct of such other Medpace Indemnitee); or
(ii) A Medpace Indemnitee shall not be entitled to indemnification hereunder to the extent such Damages are the result of a breach of any applicable federal, state or local law or a material breach of this Agreement or any Task Order by such MEDPACE Indemnitee (provided all Medpace Indemnitees not in such breach causing such Damages shall still be entitled to the indemnification hereunder notwithstanding the breach of such other Medpace Indemnitee).
14.2 Indemnification by MEDPACE
MEDPACE agrees to indemnify, defend and hold harmless VIVUS, its affiliates, and their respective officers, agents and employees, from any and all liability, loss (including reasonable attorneys’ fees) or damage any such party may suffer as the result of claims, demands, costs or judgments against them arising out of the Services to be performed pursuant to each Task Order which arise, or are alleged to, arise out of:
(i) The negligence or willful misconduct of MEDPACE; or
(ii) A breach of any applicable federal, state or local law or a material breach of this Agreement or any Task Order by MEDPACE.
15. LIMITATION OF LIABILITY
15.1. Notwithstanding the terms of Article 14 above, in no event shall VIVUS or MEDPACE be liable for any indirect, incidental, special, or consequential damages or lost profits arising out of the provision of services hereunder, even if the breaching party has been advised of the possibility of such damages unless the breaching party acted with willful misconduct in its performance of services hereunder.
16. INSPECTIONS AND AUDITS
16.1. VIVUS shall have the right, upon at least ten (10) days’ prior written notice to MEDPACE, to examine the standard operating procedures, facilities, books, records, papers, files and documentation, including computer files, data bases and records, at MEDPACE’s facilities and the facilities of clinical investigators contracted by MEDPACE to determine the adequacy of such records, to ensure the Services are being performed in accordance with the approved Task Orders and applicable regulations and/or to examine the financial records of MEDPACE as may be reasonably necessary to verify out-of-pocket expenses incurred during the performance of the Services. Such inspections and audits shall be conducted during normal business hours.
16.2. MEDPACE shall provide reasonable assistance, including making available members of its staff and providing access to all requested records, to facilitate such inspections and audits.
16.3. MEDPACE shall take all reasonable steps required by VIVUS to cure any deficiencies found in any audit, inspection or investigation.
17. DEBARMENT
17.1. MEDPACE hereby represents, warrants, and certifies that neither it nor any of its officers, directors, owners, principals or employees has been or will be at any relevant time hereunder debarred under Section 306 of the Federal Food, Drug and Cosmetic Act, 21 U.S.C. §335a(a) or (b), or similar local law. In the event that any such party becomes debarred, MEDPACE shall notify VIVUS in writing immediately.
17.2. MEDPACE hereby represents, warrants, and certifies that it has not and shall not use in any capacity the services of any individual, corporation, partnership, or association which has been debarred under Section 306 of the Federal Food, Drug and Cosmetic Act, 21 U.S.C. §335a(a) or (b), or similar local law. In the event MEDPACE becomes aware of or receives notice of the debarment of any individual, corporation, partnership, or association providing services to
MEDPACE, which relate to the Services being provided under this Agreement, MEDPACE shall notify VIVUS in writing immediately.
18. NON SOLICITATION
Neither Party and its affiliates shall during the term of this Agreement and for a period of twelve months following its termination, either directly or indirectly, hire any employee of the other Party with whom its comes into contact as a result of providing the Services, or recruit, solicit, or entice any such person to become employed by it or any affiliate and shall not approach any such employee for such purpose or encourage, authorize or approve the taking of such action by any other person. The Parties agree that any breach of this provision would cause irreparable harm and that in addition to any and all other available remedies injunctive relief, without the necessity of a bond or other security, shall be appropriate and available.
19. ENTIRE AGREEMENT
This Agreement contains the full understanding of the Parties with respect to the subject matter hereof and supersedes all existing agreements and all other oral, written or other communications between the Parties concerning the subject matter hereof. This Agreement shall not be amended, modified or supplemented in any way except in writing and signed by a duly authorized representative of VIVUS and MEDPACE.
20. GOVERNING LAW
This Agreement shall be governed by the laws of the State of California, without reference to conflicts of laws principles. In the event of breach or threatened breach, in addition to other remedies that may be available, Company shall have the right to seek specific performance and other injunctive and equitable relief, without the obligation to post bond or a security interest.
21. NO WAIVER
No waiver of any term, provision, or condition of this Agreement whether by conduct or otherwise in any one or more instances shall be deemed to be or construed as a further or continuing waiver of any such term, provisions, or conditions, or of any other term, provision, or condition of this Agreement.
22. INDEPENDENT CONTRACTOR
In fulfilling its obligations pursuant to this Agreement, each Party shall be acting as an independent contractor. Neither Party is granted any right or authority to assume or to create any obligation or responsibility, expressed or implied, on behalf of or in the name of the other Party.
23. FORCE MAJEURE
Neither Party shall be liable or deemed to be in default for any delay due to causes
beyond the reasonable control of the Party, such as: war, acts or threats of terrorism, civil disorders, acts of God, or government action; provided, that the affected Party promptly notifies the other of the cause and its effects on the Services to be performed hereunder. Financial difficulty shall never be deemed a force majeure event.
24. SEVERABILITY
In the event any provision of this Agreement shall be determined to be void or unenforceable, the remaining provisions shall remain in full force and effect.
25. ASSIGNMENT
25.1 Except as set forth herein, neither Party shall assign this Agreement or any Task Order except with the express prior written consent of the other Party.
25.2 Notwithstanding anything contained herein: (i) a Party may assign this Agreement and/or any Task Order to any Affiliate, provided that the assigning Party remains fully liable for all liabilities and obligations under this Agreement and any such Task Order; and, (ii) a Party may assign this Agreement and/or any Task Order to a Successor.
25.3 As used herein, “Affiliate means in relation to a Party, any entity controlling such Party, controlled by such Party, or under common control with such Party; and “Successor” means any entity which acquires all or substantially all assets of a Party, or all or substantially all of the assets pertaining to the subject matter of this Agreement, or any entity into which a Party is merged.
26. SUBCONTRACTING
MEDPACE may subcontract any portion of the Services to any of its Affiliates hereunder without the prior written consent of VIVUS, provided MEDPACE remains liable for the performance of any such Subcontractor.
27. CONFLICTS BETWEEN AGREEMENTS
In the event that there is any conflict between the provisions of this Agreement and any duly executed Task Order, the duly executed Task Order (but not any attachment there to) shall control.
IN WITNESS WHEREOF, the Parties have executed this Agreement as of the date first set forth above.
|
MEDPACE, INC. |
| ||
|
|
| ||
|
Signature: |
/s/ August ▇▇▇▇▇▇▇▇ |
| |
|
|
| ||
|
By: |
August ▇▇▇▇▇▇▇▇ |
| |
|
|
(Print Name) |
| |
|
|
| ||
|
Title: |
President |
| |
|
|
|
| |
|
Date: |
9/12/07 |
| |
|
|
| ||
|
VIVUS |
| ||
|
|
| ||
|
Signature: |
/s/ ▇▇▇▇▇▇ ▇. Day |
| |
|
|
| ||
|
By: |
▇▇▇▇▇▇ ▇. Day |
| |
|
|
(Print Name) |
| |
|
|
| ||
|
Title: |
V.P., Clinical Development |
| |
|
|
|
| |
|
Date: |
9/7/07 |
| |
MEDPACE
EXHIBIT A
FORM OF TASK ORDER
MEDPACE Task Order Number:
MEDPACE Project Number:
This Task Order, dated , is between Medpace, Inc. (“MEDPACE”), and VIVUS, Inc. (“VIVUS”).
RECITALS:
WHEREAS, MEDPACE and VIVUS have entered into that certain Master Services Agreement dated (the “Master Services Agreement”); and
WHEREAS, pursuant to the Master Services Agreement, MEDPACE has agreed to perform certain Services in accordance with Task Orders from time to time entered into by the Parties and VIVUS and MEDPACE now desire to enter into such a Task Order; and
WHEREAS, MEDPACE and VIVUS desire that MEDPACE provide certain services with respect to (the “Study”) for the study of the product (“Study Product”) as set out in the Protocol Number: , which is attached hereto as Appendix 1;
NOW, THEREFORE, in consideration of the mutual covenants contained herein, the Parties hereby agree as follows:
1. Scope of Work: MEDPACE shall perform the services described in the Scope of Work, attached hereto as Appendix 2, in accordance with the Project Schedule, attached hereto as Appendix 3 and any other documents attached to and specifically referenced in this Task Order (“Services”)
2. Compensation: For performance of these Services, VIVUS shall pay to MEDPACE an amount equal to the Project Budget set forth in Appendix 4, which amount shall be payable pursuant to the Payment Schedule set forth in Appendix 5.
3. Transfer of Obligations: Sponsor Obligations transferred to MEDPACE by VIVUS (consistent with the regulations set forth in 21 C.F.R. Section 312, Subpart D) are identified in Appendix 6.
4. MSA. The provisions of the Master Services Agreement are hereby expressly incorporated by reference into and made a part of this Task Order.
IN WITNESS WHEREOF, the Parties have hereunto signed this Task Order effective as of the day and year first written above.
|
MEDPACE, INC. |
| ||
|
|
| ||
|
Signature: |
|
| |
|
|
| ||
|
By: |
|
| |
|
|
(Print Name) |
| |
|
|
| ||
|
Title: |
|
| |
|
|
|
| |
|
Date: |
|
| |
|
|
| ||
|
|
| ||
|
SPONSOR |
| ||
|
|
| ||
|
Signature: |
|
| |
|
|
| ||
|
By: |
|
| |
|
|
(Print Name) |
| |
|
|
| ||
|
Title: |
|
| |
|
|
|
| |
|
Date: |
|
| |
List of Appendices:
Appendix 1: Protocol
Appendix 2: Scope of Work
Appendix 3: Project Schedule
Appendix 4: Project Budget
Appendix 5: Payment Schedule
Appendix 6: Transfer of Obligations
MEDPACE
VIVUS Task Order #01
EXHIBIT A
Qnexa OB-301 TASK ORDER
MEDPACE Task Order Number: 01
MEDPACE Project Number: VOB301
This Task Order, dated September 12, 2007, is between Medpace, Inc. (“MEDPACE”), and VIVUS, Inc. (“VIVUS”).
RECITALS:
WHEREAS, MEDPACE and VIVUS have entered into that certain Master Services Agreement dated September 12, 2007 (the “Master Services Agreement”); and
WHEREAS, pursuant to the Master Services Agreement, MEDPACE has agreed to perform certain Services in accordance with Task Orders from time to time entered into by the Parties and VIVUS and MEDPACE now desire to enter into such a Task Order; and
WHEREAS, MEDPACE and VIVUS desire that MEDPACE provide certain services with respect to a phase III, randomized, double-blind, parallel design study comparing multiple doses of VI-0521 to placebo and their single-agent phentermine and topiramate constituents for the treatment of obesity in adults (the “Study”) for the study of the product VI-0521 (“Study Product”) as set out in the Protocol Number: OB-301, which is attached hereto as Appendix 1;
NOW, THEREFORE, in consideration of the mutual covenants contained herein, the Parties hereby agree as follows:
1. Scope of Work: MEDPACE shall perform the services described in the Scope of Work, attached hereto as Appendix 2, in accordance with the Project Schedule, attached hereto as Appendix 3 and any other documents attached to and specifically referenced in this Task Order (“Services”)
2. Compensation: For performance of these Services, VIVUS shall pay to MEDPACE an amount equal to the Project Budget set forth in Appendix 4, which amount shall be payable pursuant to the Payment Schedule set forth in Appendix 5.
3. Transfer of Obligations: Sponsor Obligations transferred to MEDPACE by VIVUS (consistent with the regulations set forth in 21 C.F.R. Section 312,
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Subpart D) are identified in Appendix 6.
4. MSA. The provisions of the Master Services Agreement are hereby expressly incorporated by reference into and made a part of this Task Order.
IN WITNESS WHEREOF, the Parties have hereunto signed this Task Order effective as of the day and year first written above.
|
MEDPACE, INC. |
| ||
|
|
| ||
|
Signature: |
/s/ August ▇. ▇▇▇▇▇▇▇▇ |
| |
|
|
| ||
|
By: |
August ▇. ▇▇▇▇▇▇▇▇ |
| |
|
|
(Print Name) |
| |
|
|
| ||
|
Title: |
President |
| |
|
|
|
| |
|
Date: |
September 12, 2007 |
| |
|
|
| ||
|
|
| ||
|
SPONSOR |
| ||
|
|
| ||
|
Signature: |
/s/ ▇▇▇▇▇▇ ▇. Day |
| |
|
|
| ||
|
By: |
▇▇▇▇▇▇ ▇. Day |
| |
|
|
(Print Name) |
| |
|
|
| ||
|
Title: |
Vice President, Clinical Development |
| |
|
|
|
| |
|
Date: |
September 10, 2007 |
| |
List of Appendices:
Appendix 1: Protocol
Appendix 2: Scope of Work
Appendix 3: Project Schedule
Appendix 4: Project Budget
Appendix 5: Payment Schedule
Appendix 6: Transfer of Obligations
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
VI-0521
Protocol No. OB-301
***
CLINICAL PROTOCOL
A PHASE III, RANDOMIZED, DOUBLE-BLIND, PARALLEL-DESIGN STUDY COMPARING MULTIPLE DOSES OF VI-0521 TO PLACEBO AND THEIR SINGLE-AGENT PHENTERMINE AND TOPIRAMATE CONSTITUENTS FOR THE TREATMENT OF OBESITY IN ADULTS
|
Compound: |
|
VI-0521 |
|
|
|
|
|
Compound Name (if applicable): |
|
Phentermine plus Topiramate |
|
|
|
|
|
US IND Number (if applicable): |
|
*** |
|
|
|
|
|
Protocol Number: |
|
OB 301 |
|
|
|
|
|
Phase: |
|
3 |
|
|
|
|
|
Medical Monitor: |
|
*** |
|
|
|
|
|
Sponsor: |
|
VIVUS, Inc. |
|
|
|
|
|
Version and Date: |
|
*** |
This document contains confidential information belonging to VIVUS. Except as otherwise agreed to in writing, by accepting or reviewing this document, you agree to hold this information in confidence and not copy or disclose it to others (except where required by applicable law) or use it for unauthorized purposes. In the event of any actual or suspected breach of this obligation, VIVUS must be promptly notified.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
VIVUS — Company Confidential
INTERNAL PROTOCOL APPROVAL
|
|
|
Signature |
|
Date |
|
|
|
|
|
|
|
▇▇▇▇▇ ▇▇▇▇▇▇▇▇ |
|
|
|
|
|
Sr. Director, Clinical Research |
|
|
|
|
|
|
|
|
|
|
|
▇▇▇▇▇▇ Day, Ph.D. |
|
|
|
|
|
VP, Clinical Development |
|
|
|
|
|
|
|
|
|
|
|
▇▇▇▇▇▇▇▇▇▇ ▇▇▇▇▇▇▇▇▇, Ph.D. |
|
|
|
|
|
Sr. Director, Regulatory Affairs |
|
|
|
|
|
|
|
|
|
|
|
▇▇▇ ▇▇▇▇▇▇ |
|
|
|
|
|
▇▇. Director, Pharmaceutical Development |
|
|
|
|
PRINCIPAL INVESTIGATOR SIGNATURE
The signature below indicates that the principal investigator has read and understands the protocol and agrees to conduct the study in accordance with the protocol, applicable guidelines for Good Clinical Practices, the Declaration of Helsinki and all applicable regulatory guidelines and requirements. Please return one copy of this executed page to VIVUS, Inc.
|
Printed Name: |
|
| ||||
|
|
| |||||
|
Signature: |
|
|
Date: |
| ||
|
|
| |||||
|
Facility Name: |
|
| ||||
|
|
| |||||
|
Address: |
|
| ||||
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
PROTOCOL SYNOPSIS
Rationale:
VI-0521, a combination drug product containing phentermine hydrochloride and topiramate, has been shown in preliminary studies to result in weight loss over a prolonged period of time. These agents effect weight loss through different mechanisms, therefore, their concurrent use may provide greater efficacy than can be obtained with maximum tolerated doses of either agent alone. This study will confirm this combination drug effect by comparing weight loss associated with various doses of VI-0521, with both the individual phentermine and topiramate components comprising each combination, and with placebo.
Objectives:
The objective of this study is to evaluate the safety and efficacy of various doses of VI-0521 compared to both placebo, and the single-agent phentermine and topiramate components that comprise each combination dose. This study will provide confirmatory data to demonstrate that doses of VI-0521 have efficacy that is greater than placebo and each of the single-agent components that comprise the combination dose.
Trial Design:
In this prospective, randomized, double-blind, placebo-controlled study, eligible subjects will be randomly assigned to receive daily treatment with one of the following regimens:
***
Subjects will be enrolled at approximately *** study sites, with the intent of randomizing approximately 100 subjects into each of the *** treatment groups. The randomization will be stratified by ***.
Study treatment will consist of a 4-week titration period followed by 24 weeks of treatment. Clinic visits will occur at weeks 2 and 4 during the titration period, and subsequently every 4 weeks for the duration of treatment.
Study Subjects
Subjects included in this study will be adult men and women up to 70 years of age with BMI from *** inclusive. All female subjects who are of childbearing potential must agree to use adequate contraception, defined as a double barrier method, stable hormonal contraception plus single barrier, or tubal ligation. Major exclusions for this study include: type II diabetes; clinically significant cardiac disease; clinically significant hepatic, renal or pulmonary disease; clinically significant thyroid disease, as evidenced by signs, symptoms, or TSH >1.5 x ULN; history of bipolar disorder or psychosis, depression of moderate or greater severity, or presence or history of suicidal behavior or active suicidal ideation; obesity of known genetic or endocrine
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
origin; recent history of weight instability, or recent participation in a formal weight loss program; history of glaucoma; and smoking cessation within 3 months prior to study enrollment.
Efficacy Endpoints:
The primary study endpoints will be based on the percent weight loss at week 28, calculated as ***, and the percentage of subjects achieving at least 5% weight loss at week 28. For subjects who discontinue treatment prior to study completion, every attempt will be made to have them return for week 28 evaluations. ***.
Secondary efficacy endpoints will include the proportion of subjects achieving 10% weight loss, change from baseline in waist circumference, and change from baseline in *** and *** at ***. As was done for the primary endpoints, subjects who discontinue prior to study completion ***.
Additional endpoints will include *** assessments of *** and ***, changes in primary and secondary outcomes over monthly intervals during the study, and Framingham 10-year risk assessments.
*** will also be evaluated. Data will be obtained using a multiple trough sampling scheme with samples collected at *** and *** from each subject. Effects of various cofactors including (but not limited to) ***, gender, race, ***, and age will be evaluated.
Safety Endpoints
Safety endpoints will include adverse events, ***. Adverse event assessments will include direct questions related to eye pain and *** will be assessed at each visit using the ***.
Statistical Methods:
Comparisons between treatments will be assessed using a *** with factors of *** and ***, and with ***. A step down multiple comparison procedure will be utilized to protect overall ***. For the first level of testing, the response to the *** will be compared to ***, and to ***. If each of these *** comparisons is significant at the *** for both of the co-primary endpoints, then this combination will be considered to have met requirements, and testing will continue to the second level, which will evaluate the *** compared to *** and ***. If all comparisons at the second level are met at the ***, differences in treatment effect between the *** and the ***, the ***, and the *** will be presented using ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
TABLE OF CONTENTS
|
INTERNAL PROTOCOL APPROVAL |
2 |
|
|
|
|
PRINCIPAL INVESTIGATOR SIGNATURE |
2 |
|
|
|
|
PROTOCOL SYNOPSIS |
3 |
|
|
|
|
TABLE OF CONTENTS |
5 |
|
|
|
|
1. INTRODUCTION |
11 |
|
|
|
|
1.1. Background |
11 |
|
|
|
|
1.2. Rationale |
12 |
|
|
|
|
2. TRIAL OBJECTIVES |
12 |
|
|
|
|
3. TRIAL DESIGN |
12 |
|
|
|
|
4. SUBJECT SELECTION |
13 |
|
|
|
|
4.1. Inclusion Criteria |
13 |
|
|
|
|
4.2. Exclusion Criteria |
14 |
|
|
|
|
4.3. Randomization Criteria |
15 |
|
|
|
|
4.4. Life Style Guidelines |
16 |
|
|
|
|
5. TRIAL TREATMENTS |
16 |
|
|
|
|
5.1. Allocation to Treatment |
16 |
|
|
|
|
5.2. Breaking the Blind |
16 |
|
|
|
|
5.3. Drug Supplies |
17 |
|
|
|
|
5.3.1. Formulation and Packaging |
17 |
|
|
|
|
5.3.2. Preparation and Dispensing |
17 |
|
|
|
|
5.3.3. Administration |
18 |
|
|
|
|
5.3.4. *** |
18 |
|
|
|
|
5.3.5. Compliance |
18 |
|
|
|
|
5.4. Drug Storage and Drug Accountability |
18 |
|
|
|
|
5.5. Concomitant Medication(s) |
18 |
|
|
|
|
5.5.1. Excluded Medications |
18 |
|
|
|
|
5.5.2. Other Restricted Medications |
19 |
|
|
|
|
5.5.3. Documentation of Concomitant Medication Use |
19 |
|
|
|
|
6. TRIAL PROCEDURES |
20 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
6.1. Schedule of Visits |
20 |
|
|
|
|
6.2. Trial Period |
25 |
|
|
|
|
6.3. Subject Withdrawal |
25 |
|
|
|
|
7. ASSESSMENTS |
26 |
|
|
|
|
7.1. Weight and Waist Measurement Assessments |
26 |
|
|
|
|
7.2. ▇▇▇▇▇ ▇▇▇▇▇ |
27 |
|
|
|
|
7.3. Questionnaires |
27 |
|
|
|
|
7.3.1. *** |
27 |
|
|
|
|
7.3.2. *** |
28 |
|
|
|
|
7.3.3. *** |
28 |
|
|
|
|
7.4. *** |
28 |
|
|
|
|
7.5. *** |
28 |
|
|
|
|
7.6. Laboratory Tests |
28 |
|
|
|
|
7.7. Physical Examination |
30 |
|
|
|
|
7.8. Electrocardiograms (ECG) |
30 |
|
|
|
|
8. ADVERSE EVENT REPORTING |
30 |
|
|
|
|
8.1. Adverse Events |
30 |
|
|
|
|
8.1.1. Severity Assessment |
31 |
|
|
|
|
8.1.2. Causality Assessment |
31 |
|
|
|
|
8.1.3. Abnormal Test Findings |
31 |
|
|
|
|
8.2. Serious Adverse Events |
32 |
|
|
|
|
8.2.1. Definition of Hospitalization |
32 |
|
|
|
|
8.3. Eliciting Adverse Event Information |
33 |
|
|
|
|
8.3.1. Eye Pain |
33 |
|
|
|
|
8.3.2. *** |
33 |
|
|
|
|
8.4. Reporting Period |
33 |
|
|
|
|
8.5. Reporting Requirements |
34 |
|
|
|
|
8.5.1. Serious Adverse Event Reporting Requirements |
34 |
|
|
|
|
8.5.2. Non-Serious Adverse Event Reporting Requirements |
34 |
|
|
|
|
8.5.3. *** |
34 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
9. DATA ANALYSIS/STATISTICAL METHODS |
35 |
|
|
|
|
9.1. Sample Size Determination |
35 |
|
|
|
|
9.2. Efficacy Analysis |
35 |
|
|
|
|
9.2.1. Analysis of Primary Endpoint |
35 |
|
|
|
|
9.2.2. Analysis of Secondary Endpoints |
36 |
|
|
|
|
9.3. Analysis of Other Endpoints |
36 |
|
|
|
|
9.4. Safety Analysis |
36 |
|
|
|
|
9.5. Interim Analysis |
37 |
|
|
|
|
9.6. *** |
37 |
|
|
|
|
10. QUALITY CONTROL AND QUALITY ASSURANCE |
37 |
|
|
|
|
11. DATA HANDLING AND RECORD KEEPING |
37 |
|
|
|
|
11.1. Case Report Forms / Electronic Data Record |
37 |
|
|
|
|
11.2. Record Retention |
38 |
|
|
|
|
12. ETHICS |
38 |
|
|
|
|
12.1. Institutional Review Board (IRB)/Independent Ethics Committee (IEC) |
38 |
|
|
|
|
12.2. Ethical Conduct of the Trial |
39 |
|
|
|
|
12.3. Subject Information and Consent |
39 |
|
|
|
|
12.4. Disclosure of Data |
39 |
|
|
|
|
13. REGULATORY CORRESPONDENCE |
39 |
|
|
|
|
14. DEFINITION OF END OF TRIAL (IF APPLICABLE) |
40 |
|
|
|
|
15. SPONSOR DISCONTINUATION CRITERIA |
40 |
|
|
|
|
16. PUBLICATION OF TRIAL RESULTS |
40 |
|
|
|
|
17. REFERENCES |
42 |
|
|
|
|
APPENDIX 1: SCHEDULE OF STUDY ACTIVITIES |
44 |
|
|
|
|
APPENDIX 2: *** |
45 |
|
|
|
|
APPENDIX 3: *** |
47 |
|
|
|
|
APPENDIX 4: PROTOCOL AMENDMENTS |
50 |
|
LIST OF TABLES |
|
|
|
|
|
Table 1. Dosage strengths by titration week for each treatment group |
17 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
LIST OF FIGURES |
|
|
|
|
|
Figure 1. Schematic representation of study design. |
13 |
|
|
|
|
Figure 2. Measuring Tape Position for Waist Circumference Assessments |
27 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
LIST OF ABBREVIATIONS
|
mU |
|
Micro units |
|
ACE |
|
Angiotensin Converting Enzyme |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
BMI |
|
Body Mass Index |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
CFR |
|
Code of Federal Regulations |
|
CNS |
|
Central Nervous System |
|
CO2 |
|
Carbon Dioxide |
|
CRF |
|
Case Report Form |
|
*** |
|
*** |
|
dL |
|
deciliter |
|
ECG |
|
Electrocardiogram |
|
eg |
|
For example |
|
FDA |
|
Food and Drug Administration |
|
*** |
|
*** |
|
HBsAg |
|
Hepatitis B Surface Antigen |
|
HCV |
|
Hepatitis C Virus |
|
HDL |
|
High Density Lipoprotein |
|
HIV |
|
Human Immunodeficiency Virus |
|
I-CAM |
|
Intercellular Cell Adhesion Molecule |
|
ICH-GCP |
|
International Committee for the Harmonization of Good Clinical Practices |
|
IEC |
|
Independent Ethics Committee |
|
IND |
|
Investigational New Drug |
|
IRB |
|
Institutional Review Board |
|
IVRS |
|
Interactive Voice Response System |
|
*** |
|
*** |
|
kcal |
|
Kilocalorie |
|
kg |
|
Kilogram |
|
*** |
|
*** |
|
LDL |
|
Low Density Lipoprotein |
|
LOCF |
|
Last Observation Carried Forward |
|
m |
|
Meter |
|
mg |
|
Milligram |
|
mL |
|
Milliliter |
|
mmHg |
|
Millimeters of Mercury |
|
NYHA |
|
New York Heart Association |
|
PAI-1 |
|
Plasminogen Activator Inhibitor 1 |
|
*** |
|
*** |
|
QoL |
|
Quality of Life |
|
RBP-4 |
|
Retinol Binding Protein 4 |
|
*** |
|
*** |
|
*** |
|
*** |
|
TSH |
|
Thyroid Stimulating Hormone |
|
UA |
|
Urinalysis |
|
*** |
|
*** |
|
V-CAM |
|
Vascular Cell Adhesion Molecule |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
WNL |
|
Within Normal Limits |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
1. INTRODUCTION
Phentermine hydrochloride is a synthetic sympathomimetic amine that is approved by the FDA for the short-term treatment of obesity.(1) The usual adult dosage of phentermine hydrochloride is *** administered either once daily or in divided doses. Topiramate is a sulfamate-substituted monosaccharide that is approved by the FDA for the treatment of partial onset seizures, primary generalized tonic-clonic seizures, seizures associated with Lennox-Gastaut syndrome, and for migraine headache prophylaxis. Topiramate has been shown to ***.(2) The recommended total daily dose of topiramate for treatment of seizures in adults is *** administered in two divided doses. The present study is being conducted to evaluate the combined use of phentermine at doses up to 15 mg/day, and topiramate at doses up to 92 mg/day for the treatment of obesity.
1.1. Background
It is estimated that approximately 129 million adults in the United States are clinically overweight or obese.(3) In recent years, there has been a dramatic increase in obesity in both children and adults.(4),(5) Results from the National Health and Nutrition Examination Survey showed an overall 32.2% prevalence of obesity during 2003-2004, with obesity increasing from 27.5% in 1999-2000 to 33.4% in 2003-2004 in men but remaining relatively unchanged in women (1999-2000, 33.4%; 2003-2004, 33.2%).(5)
Obesity is associated with numerous co-morbidities including dyslipidemia, coronary artery disease (CAD), hypertension, stroke and type 2 diabetes.(4),(6) Epidemiological data indicate that obesity is associated with increased mortality,(7) and a recent study of over 500,000 individuals concluded that excess body weight during midlife, including overweight, was associated with an increased risk of death.(8) A modest weight loss (5-10%) can result in a marked reduction in obesity-related metabolic and cardiovascular risk factors.(9),(10),(11) Diet, exercise and behavior modification are standard treatments for obesity, however most obese individuals do not achieve prolonged weight reduction without supplemental pharmacotherapy. Medications currently approved by FDA for weight loss are often poorly tolerated due to side effects and often fail to maximize long-term efficacy.(12)
Phentermine hydrochloride, a synthetic sympathomimetic amine, is an anorectic agent approved by the FDA as a short-term adjunct to a weight loss regimen based on exercise, behavior modification and caloric restriction. The mechanism of action of phentermine for weight loss is similar to that of other anorectic agents; it has peripheral sympathomimetic actions and stimulates the central nervous system.(1) It is postulated that ***, and there is a ***.(13),(14) Additionally, increased *** levels may result in a decrease in *** that may result in increased satiety and decreased appetite.
Topiramate, a sulfamate-substituted monosaccharide, is an antiepileptic agent indicated as adjunctive therapy for partial onset seizures, primary generalized tonic-clonic seizures, seizures associated with Lennox-Gastaut syndrome and for migraine headache prophylaxis.(2) The recommended total daily dose of topiramate for treatment of seizures in adults is ***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
administered in two divided doses; ***. Topiramate is known to ***.(2) Recent clinical studies have shown that topiramate may promote weight loss in ***.(15),(16),(17) However, the exact mechanism by which topiramate exerts its anorectic effect is unknown.
VI-0521 has been studied in a Phase 2, randomized, double-blind clinical trial of 200 otherwise healthy obese adults under an Investigator-Initiated IND.(18) In this study, subjects were randomized into 1 of 4 treatment groups: ***. Treatment was continued for ***. Weight loss among subjects treated with VI-0521 ***, compared to *** among *** among ***, and *** among ***. Subjects treated with VI-0521 *** than subjects in the other treatment groups. Significant decreases vs. *** and *** were also observed in subjects receiving VI-0521. Of the 200 subjects randomized, *** completed treatment to ***, and a *** completion rate was reported for subjects in the *** group. No deaths or serious adverse events were reported during the study and no significant changes in heart valve morphology were observed. The most commonly reported adverse events in subjects treated with VI-0521 were ***.
1.2. Rationale
VI-0521 is an investigational weight loss therapy that is a combination of two currently approved drugs, phentermine and topiramate. As such, VI-0521 represents a potential advance in the medical treatment of obesity since the two agents comprising this combination product effect weight loss through different mechanisms. Additionally, some of the expected side effects of the two drugs may be mitigated by complementary effects of the other. Thus, combining phentermine with topiramate may produce a similar or better adverse event profile compared to either of these agents individually. Topiramate therapy has been associated with ***. Due to the ***, it is mechanistically possible that ***. Thus, the dual mechanisms and low drug doses employed in VI-0521 may provide a safe and effective pharmacotherapy for the achievement and maintenance of weight loss in obese adults.
2. TRIAL OBJECTIVES
The primary objective of this trial is to demonstrate that two different dose levels of VI-0521, a fixed combination of phentermine and topiramate, result in weight loss that is greater than both placebo, and the single agent phentermine and topiramate constituents that comprise each combination dose. Additional study objectives are to evaluate the safety of combination doses compared to both placebo and their single-agent constituents.
3. TRIAL DESIGN
In this prospective, randomized, double-blind, placebo and single agent-controlled trial, subjects meeting the eligibility criteria will be randomly assigned (with equal probability) among the *** treatment groups described in Figure 1.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
Figure 1. Schematic representation of study design.
Subjects will be recruited at approximately *** study sites with the intent of randomizing 100 subjects into each of the *** treatment groups. The randomization schedule will be stratified by *** to ***.
The first 4 weeks of drug treatment will be a titration period, during which doses will be gradually increased at *** intervals until the specified dose is reached. The 4-week titration period will be followed by an additional 24 weeks of drug treatment at the assigned dose level. Clinic visits will occur at the end of weeks 2 and 4 during the titration period, and at 4-week intervals thereafter.
The primary efficacy endpoints for this trial will be based on the percent weight loss at week 28, calculated as ***, and the percentage of subjects achieving at least 5% weight loss at week 28. Secondary efficacy endpoints will include the percentage of subjects losing 10% of their body weight, reductions in waist circumference, and changes in ***. For each of these endpoints, statistical comparisons will evaluate differences between *** and ***, and between *** and the *** doses that comprise each combination. Additional efficacy variables will include *** of *** and ***, and changes in primary and secondary outcomes over monthly intervals during the trial, and Framingham 10-year risk assessments.(19)
*** will also be obtained, and effects of various cofactors including (but not limited to) ***, gender, race, ***, and age will be evaluated. ***.
Safety evaluations will include summaries of adverse events, ***.
4. SUBJECT SELECTION
This clinical trial can fulfill its objectives only if appropriate subjects are enrolled. The following eligibility criteria are designed to select subjects for whom protocol treatment is considered appropriate. All relevant medical and non-medical conditions should be taken into consideration when deciding whether this protocol is suitable for a particular subject.
4.1. Inclusion Criteria
To be eligible for enrollment into this trial, subjects must meet all of the following criteria. Specifically, subjects must:
1. Be adults 70 years of age or less with a BMI between ***;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
2. If females of child-bearing potential, be using adequate contraception, defined as double barrier methods, stable hormonal contraception plus single barrier, or tubal ligation. Females are considered to be of child-bearing potential unless they are at least 55 years of age with spontaneous amenorrhea for at least 12 months or have a documented FSH value of 40 IU/L or greater, or have had a hysterectomy or bilateral oophorectomy;
3. Provide written informed consent;
4. Be willing and able to comply with scheduled study visits, treatment plan, laboratory tests, and other study procedures;
4.2. Exclusion Criteria
Subjects presenting with any of the following will not be included in the trial:
1. Known allergy or hypersensitivity to phentermine or topiramate, or use of phentermine or topiramate for any indication within the past 3 months;
2. Weight gain or loss of greater than 5 kg, use of a very low-calorie diet, or participation in a formal weight loss program (investigational or otherwise) within the past 3 months (this includes: Weight Watchers and related dietary/lifestyle intervention programs; prepared food programs; prescribed or over-the-counter weight loss medications; dietary supplement or herbal preparations, teas, or tinctures intended for weight loss; or any supervised fast or very low calorie diet);
3. Obesity that is of a known genetic or endocrine origin;
4. History of eating disorders (eg. bulimia, binge eating disorder), drug abuse, or alcohol abuse (defined as >14 drinks per week) within the past year;
5. Previous bariatric surgery;
6. Smoking cessation within the previous 3 months or plans to quit smoking during study participation;
7. History of glaucoma or any past or present use of medications to treat increased intraocular pressure;
8. Clinically significant thyroid dysfunction as evidenced by signs or symptoms of hypothyroidism, a TSH > 1.5 x ULN, or use of thyroid hormone treatment that has not been stable for at least 3 months;
9. Use of chronic systemic glucocorticoid therapy, or any other steroid hormone therapy that has not been stable for at least 3 months;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
10. Any history of bipolar disorder or psychosis, greater than one lifetime episode of major depression, current depression of moderate or greater severity (PHQ-9 score of 10 or more), presence or history of suicidal behavior or ideation with some intent to act on it, or antidepressant use that has not been stable for at least 3 months;
11. Diagnosis of type 2 diabetes by history, or as confirmed by a fasting blood glucose of 126 mg/dL or greater, or history of any antidiabetic medication use;
12. Stroke, myocardial infarction, life-threatening arrhythmia, or coronary re-vascularization within the past 6 months;
13. Unstable angina, NYHA class II-IV congestive heart failure, or known clinically significant cardiac valvulopathy;
14. Systolic blood pressure greater than 160 mmHg, or diastolic blood pressure greater than 100 mmHg;
15. Cholelithiasis within the past 6 months;
16. Any history of nephrolithiasis;
17. History of malignancy within the past 5 years other than basal or squamous cell carcinomas of the skin that have been completely excised, or cervical cancers that have been surgically removed;
18. Pregnancy, breastfeeding, or plans for pregnancy during the study period;
19. Use of any investigational medication or device for any indication within the last month;
20. Evidence of any clinically significant renal, hepatic, pulmonary, psychiatric, or other condition that, in the opinion of the investigator, would contraindicate the administration of study medications, interfere with study evaluations, or confound the interpretation of study results.
4.3. Randomization Criteria
The following additional criteria must be met prior to dispensing treatment to trial subjects:
1. Baseline physical examination, ECG, and laboratory findings with no abnormalities that are considered clinically significant by the principal investigator;
2. Laboratory values that are within the ranges specified below:
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
4.4. Life Style Guidelines
Prior to randomization, subjects will be counseled on how to reduce their caloric intake by 500 kcal/day, and on the importance of light daily exercise. In order to facilitate discussions between study subjects and research staff, subjects will complete a 24-hour dietary recall at their randomization visit, however, information provided as part of this exercise will be used only for discussions related to recommended dietary modification.
Subjects randomized into the study will also be advised to initiate a lifestyle change program utilizing the LEARN® Program for Weight Management. The LEARN® program is a 12-week program designed to aid in weight management by providing tools to facilitate lifestyle, attitude, relationship, nutrition and exercise changes. Each subject will be provided with a LEARN® manual and advised to read and implement the material as appropriate to their individual situation. Site personnel will be encouraged to discuss these materials with subjects at their regularly scheduled visits, however, no data will be collected to document the level of compliance with the program’s dietary, lifestyle and/or exercise recommendations.
5. TRIAL TREATMENTS
5.1. Allocation to Treatment
Subjects will be assigned to study treatment using a centrally managed, computer-generated randomization schedule. This randomization will be stratified by gender, and will assign subjects among the *** treatment groups with equal probability.
To implement the randomization of subjects among treatment groups, each participating site will be pre-stocked with titration kits corresponding to each treatment group. When study subjects qualify for randomization at the appropriate study visit (see Section 6), site personnel will contact an Interactive Voice Response System (IVRS), either by telephone or through a designated website, and provide the requested information about the study subject. The randomization assignment will then be made, and the site will be instructed to dispense a specific kit number to the study subject. Additional kits will then be shipped to sites to replace those dispensed to subjects according to the randomization schedule. When subjects return for subsequent study visits, the IVRS system will be used to dispense treatment kits.
5.2. Breaking the Blind
Blinding of drug kits must not be broken during the study unless it considered necessary by the investigator for the management of an adverse event or other medical emergency. In the event of such medical emergency, the identity of the study treatment is obtained by contacting the IVRS. Should unblinding of any subject’s treatment occur, VIVUS would be notified ***. Investigators are also required to ensure that any potential serious adverse events are reported according to the requirements outlined in Section 8.2, and to send a written report to VIVUS within *** days to
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
document the reason for unblinding. Should unblinding of any subject’s treatment occur, the subject must discontinue trial participation.
5.3. Drug Supplies
5.3.1. Formulation and Packaging
Medications for this trial will consist of ***. Doses specified for each treatment group will be achieved by varying the *** added to each capsule. Regardless of the dosage assignment, all study treatments will be administered as a ***.
Clinical supplies will be manufactured for VIVUS by *** in accordance with current Good Manufacturing Practices. All clinical supplies will be labeled with information required by national and/or international regulations. Study drug will be packaged into 2 types of kits, titration kits for use during the first 4 weeks of study therapy when doses are being increased gradually to the final assigned dose, and treatment kits, for use once subjects have been titrated to their assigned dosage of medication. Titration kits consist of a ***. Each *** on the *** will be labeled with the *** and will contain capsules with the dose specified for that week of treatment, as outlined in Table 1.
Table 1. Dosage strengths by titration week for each treatment group.
|
|
|
|
|
*** |
| ||||||
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
Titration kits will be labeled with the ***. *** will be labeled with the ***.
Treatment kits will consist of bottles, *** of study medication at the treatment dosages shown in Table 1. Each kit will contain a single bottle of capsules, will be labeled with the ***.
5.3.2. Preparation and Dispensing
Clinical supplies provided by the sponsor are to be dispensed only by or under the direct supervision of qualified investigators to subjects meeting the criteria for study entry and in accordance with this protocol. Assignment of specific drug kits to study subjects will require the use of the IVRS system, however, no other preparation of clinical supplies is required of the investigational staff.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
5.3.3. Administration
Investigators will instruct subjects to take 1 capsule of study medication every morning. When dispensing titration kits, investigators should ensure that subjects understand that each card contains a 4-week supply of medication, and that the capsules must be taken ***.
5.3.4. ***
Should *** would cause subjects to consider ***, investigators may, at their discretion, ***. *** are possible with agreement from the medical monitor. Such events may or may not be related to trial treatment.
All subjects undergoing *** for ***may be *** based on discretion of the PI. If dosing has been ***, a new titration kit should be ordered through *** to ***. For shorter ***, subjects may ***. All subjects whose trial treatment has been *** will be encouraged to *** and to attend their regularly scheduled study visits.
5.3.5. Compliance
Subject compliance with trial medication will be assessed by ***, and *** should plan any corrective action necessary. Subjects who remain noncompliant with study dosing despite corrective actions by site personnel may be discontinued from the trial.
5.4. Drug Storage and Drug Accountability
All unused study drug must be stored in its packaging at room temperature in a dry, secure area. Access to drug storage areas should be limited to the investigator and staff involved with the study. All used and unused drug must be maintained at the study site, and made available for audit by VIVUS, Inc. personnel.
The investigator must maintain records documenting the amount, condition, and date of delivery of all study drug received from the Sponsor. In addition, all drug dispensed to study subjects during the course of the study must be ***. Subjects must be instructed to *** by each subject. No investigational drug, used or unused, may be discarded. All used and unused drug must be returned to the sponsor or designated representative upon completion of the study.
5.5. Concomitant Medication(s)
5.5.1. Excluded Medications
Subjects must not take the following medications during their participation in this trial:
· ***;
· ***;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· ***;
· ***;
· ***.
Although the need for any antidiabetic medications at screening would exclude subjects from trial participation, subjects who develop a need for these medications during the course of the trial need not be discontinued. Should antidiabetic medications be required by any trial subjects, it is recommended that *** be used initially, followed by *** and/or ***. ***, either alone or in combination with other medications, should be reserved for subjects who cannot achieve adequate control with other modes of treatment. *** are prohibited, and subjects requiring treatment with these medications must be discontinued from the trial. Subjects whose *** cannot be adequately controlled with the concomitant treatments allowed in this trial should be discontinued and referred back to their primary health care provider for further follow-up (see Section 6.3).
5.5.2. Other Restricted Medications
Subjects using *** must be on doses that have been stable for at least 3 months. For subjects who develop symptoms consistent with *** during the study, *** replacement may be initiated following appropriate diagnostic workup. Similarly, subjects whose needs for *** medications change may have these treatments added or changed as clinically indicated. In the event that subjects require any other changes in these medications, the Sponsor should be contacted regarding their continued eligibility.
All medications used for the treatment of *** associated with *** must be stable for at least 1 month prior to trial entry; however, adjustment of these medications during the trial is permitted if subjects’ requirements for treatment change. For subjects whose ***, or who exhibit symptoms associated with *** during the trial, *** agents should be withdrawn or doses should be reduced. For this trial, it is recommended that *** be the first medications to be reduced or withdrawn followed by ***.
For subjects whose ***, *** therapy should be initiated with *** or ***. If these medications are already present, *** may be added.
Subjects whose *** cannot be adequately controlled with the concomitant treatments allowed in this trial should be discontinued and referred back to their primary health care provider for further follow-up (see Section 6.3).
5.5.3. Documentation of Concomitant Medication Use
All concomitant medications, including over-the-counter products and nutritional/herbal supplements, must be listed on the appropriate case report form at trial entry. Any changes in concomitant medication during the course of the trial must also be noted on the appropriate CRF.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
6. TRIAL PROCEDURES
6.1. Schedule of Visits
A schedule of study activities by visit is shown in Appendix 1. A detailed list of activities conducted at each study visit is also described in the following section.
VISIT 1: Screening:
· Obtain written informed consent;
· Obtain medical history;
· Assess ▇▇▇▇▇ ▇▇▇▇▇, weight, height, waist circumference, and BMI;
· Administer ***, and ***;
· Assess inclusion/exclusion criteria;
· Obtain blood and urine samples for laboratory analyses, and perform urine pregnancy test (female subjects of childbearing potential only);
· Schedule a follow-up visit in 2 weeks (±1 week).
VISIT 2: Randomization: Week 0:
· Perform urine pregnancy test (female subjects of childbearing potential only);
· Perform physical examination including ECG evaluation (can be done at any time between screening;
· If screening laboratory results and physical examination findings are acceptable, obtain randomization assignment through IVRS;
· Assess weight, waist circumference, and ▇▇▇▇▇ ▇▇▇▇▇;
· Administer *** for *** and ***, and perform *** (see Section 7.4);
· Perform 24-hour dietary recall with subject and review lifestyle modifications;
· Dispense assigned titration kit and provide instructions for proper use;
· Assess adverse events (including eye pain), and changes in concomitant medications;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Schedule next follow-up visit in 2 weeks (±2 days).
VISIT 3: Titration: Week 2:
· Perform urine pregnancy test (female subjects of childbearing potential only);
· Assess weight, waist circumference, and ▇▇▇▇▇ ▇▇▇▇▇;
· Administer *** and ***;
· Administer *** for *** and ***;
· Review lifestyle modifications with subject;
· Review assigned titration kit to assess medication use and treatment compliance. Re-dispense kit and provide proper instructions for continued use;
· Assess adverse events (including eye pain), and changes in concomitant medications;
· Schedule next follow-up visit in 2 weeks (±2 days).
VISIT 4: Titration: Week 4:
· Collect blood sample for laboratory tests (all subjects) and perform urine pregnancy test (female subjects of childbearing potential only);
· Assess weight, waist circumference, and ▇▇▇▇▇ ▇▇▇▇▇;
· Administer *** and ***;
· Administer *** for *** and ***, and perform *** (see Section 7.4);
· Review lifestyle modifications with subject;
· Collect titration kit, assess treatment compliance, and perform drug accountability;
· Obtain treatment kit assignment through IVRS, dispense kit to subject, and provide proper instructions for use;
· Assess adverse events (including eye pain), and changes in concomitant medications;
· Schedule next follow-up visit in 4 weeks (±1 week).
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
VISIT 5: Treatment: Week 8:
· Collect blood sample for laboratory tests (all subjects), and perform urine pregnancy test (female subjects of childbearing potential only);
· Assess weight, waist circumference, and ▇▇▇▇▇ ▇▇▇▇▇;
· Administer *** and ***;
· Administer *** for *** and ***;
· Review lifestyle modifications with subject;
· Collect used treatment kit, assess treatment compliance, and perform drug accountability;
· Obtain treatment kit assignment through IVRS, dispense kit to subject, and provide proper instructions for use;
· Assess adverse events (including eye pain), and changes in concomitant medications;
· Schedule next follow-up visit in 4 weeks (±1 week).
VISIT 6: Treatment: Week 12:
· Perform urine pregnancy test (female subjects of childbearing potential only);
· Assess weight, waist circumference, and ▇▇▇▇▇ ▇▇▇▇▇;
· Administer *** and ***;
· Administer *** for *** and ***;
· Review lifestyle modifications with subject;
· Collect used treatment kit, assess treatment compliance, and perform drug accountability;
· Obtain treatment kit assignment through IVRS, dispense kit to subject, and provide proper instructions for use;
· Assess adverse events (including eye pain), and changes in concomitant medications;
· Schedule next follow-up visit in 4 weeks (±1 week). Subjects should be reminded not to take their study medication on the morning of their next visit, but instead, to bring it to
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
the visit with them, and take it after blood samples have been obtained. Subjects should also note the time of their last dose prior to the next visit.
VISIT 7: Treatment: Week 16
· Ensure that the previous dose of trial medications was taken between 20 and 28 hours ago, and obtain a *** blood sample for *** evaluations (if the previous dose of trial medications is outside this window, *** should be postponed for 1 day);
· Collect blood sample for laboratory tests (all subjects), and perform urine pregnancy test (female subjects of childbearing potential only);
· Assess weight, waist circumference, and ▇▇▇▇▇ ▇▇▇▇▇;
· Administer *** and ***;
· Administer *** for *** and ***;
· Review lifestyle modifications with subject;
· Collect used treatment kit, assess treatment compliance, and perform drug accountability;
· Obtain treatment kit assignment through IVRS, dispense kit to subject, and provide proper instructions for use;
· Assess adverse events (including eye pain), and changes in concomitant medications;
· Schedule next follow-up visit in 4 weeks (±1 week).
VISIT 8: Treatment: Week 20:
· Perform urine pregnancy test (female subjects of childbearing potential only);
· Assess weight, waist circumference, and ▇▇▇▇▇ ▇▇▇▇▇;
· Administer *** and ***;
· Administer *** for *** and ***;
· Review lifestyle modifications with subject;
· Collect used treatment kit, assess treatment compliance, and perform drug accountability;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Obtain treatment kit assignment through IVRS, dispense kit to subject, and provide proper instructions for use;
· Assess adverse events (including eye pain), and changes in concomitant medications;
· Schedule next follow-up visit in 4 weeks (±1 week).
VISIT 9: Treatment: Week 24:
· Perform urine pregnancy test (female subjects of childbearing potential only);
· Assess weight, waist circumference, and ▇▇▇▇▇ ▇▇▇▇▇;
· Administer *** and ***;
· Administer *** for *** and ***;
· Review lifestyle modifications with subject;
· Collect used treatment kit, assess treatment compliance, and perform drug accountability;
· Obtain treatment kit assignment through IVRS, dispense kit to subject, and provide proper instructions for use;
· Assess adverse events (including eye pain), and changes in concomitant medications;
· Schedule next follow-up visit in 4 weeks (±1 week). Subjects should be reminded not to take their trial medication on the morning of their next visit, but instead, to bring it to the visit with them, and take it after blood samples have been obtained. Subjects should also note the time of their last dose prior to the next visit.
VISIT 10: End of Treatment: Week 28 (or early term):
· Ensure that the previous dose of trial medications was taken between 20 and 28 hours ago, and obtain a *** blood sample for *** evaluations (if the previous dose of trial medications is outside this window, *** should be postponed for 1 day);
· Collect blood and urine samples for laboratory tests (all subjects), and perform urine pregnancy test (female subjects of childbearing potential only);
· Assess weight, waist circumference, and ▇▇▇▇▇ ▇▇▇▇▇;
· Administer ***, and ***;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Perform physical examination including ECG evaluation;
· Administer *** for *** and ***, and perform *** (see Section 7.4);
· Collect used treatment kit, assess treatment compliance, and perform drug accountability;
· Assess adverse events (including eye pain), and changes in concomitant medications;
· Ask End of Treatment Questions (see Section 7.5);
· Discontinue subject’s trial participation.
6.2. Trial Period
For each subject, the trial period will begin when written informed consent is provided and will continue until Visit 10 (Week 28 or early termination) is completed. In certain instances, however, adverse event information may be required for events that occur after the trial period (see Section 8).
6.3. Subject Withdrawal
Subjects may withdraw from the trial at any time and for any reason. Additionally, the investigator must discontinue participation for any subject who:
· Experiences an adverse event that, based on the investigator’s medical judgment, would jeopardize the subject’s well being or prevent the safe completion of the study;
· Becomes pregnant;
· Requires other medical treatment that is excluded by the protocol;
· Has their treatment unblinded by the investigator;
· Is unwilling to comply with the provisions of the protocol.
Investigators must discontinue trial participation for all subjects if the Sponsor terminates the trial. Evaluations that would normally be performed upon trial completion should be made for all subjects who exit the trial prematurely. For subjects who have received ***, this includes obtaining appropriate samples for laboratory testing, and performing a physical examination (including ECG). Additionally, for all subjects who discontinue trial participation prematurely, investigators should make all reasonable attempts to schedule a clinic visit at week 28 to make a final weight assessment.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
If a subject does not return for a scheduled visit, every effort should be made to contact the subject. In any circumstance, every effort should be made to document subject outcome, if possible. The investigator should ***
If the subject withdraws from the trial and also withdraws consent for disclosure of future information, ***. ***.
7. ASSESSMENTS
7.1. Weight and Waist Measurement Assessments
Subjects should be weighed using a calibrated digital scale. The same scale should be used for each measurement and measurements should be evaluated by the same site personnel at each visit, whenever possible. Subject weights should be obtained, whenever possible, under the same conditions (no shoes, clothing of similar weight) that were employed at the first (screening) weighing. Subjects should be encouraged to complete their weigh-in visits in the morning and should be fasting prior to weigh-in.
Waist measurement will be performed using a measuring tape provided by VIVUS, Inc., and should be obtained by the same individual at each visit. To measure the waist circumference, locate the top of the right iliac crest, and place the measuring tape in a horizontal plane (parallel to the floor) around the abdomen at the level of the top of the iliac crest, as shown in Figure 2.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
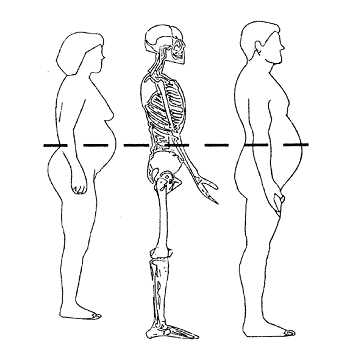
Figure 2. Measuring Tape Position for Waist Circumference Assessments
Ensure that the subject is relaxed. Ensure that the tape is snug but does not indent or compress the skin, and make the measurement at the end of a normal expiration.
7.2. ▇▇▇▇▇ ▇▇▇▇▇
▇▇▇▇▇ ▇▇▇▇▇ including blood pressure, pulse rate, temperature, and respiration rate will be assessed at each study visit. All blood pressure measurements should be obtained after subjects have been resting in the seated position for at least ***. Pulse rate and respiratory rate measurements should be made by counting events (heartbeats or breaths) for a period of 30 seconds and multiplying these values by 2 to obtain the rates per minute. Whenever possible, the same person should do all of the assessments for a given subject.
7.3. Questionnaires
7.3.1. ***
The *** that is to be completed ***, and ***. This questionnaire is designed to evaluate the ***.(20),(21) Because this instrument is intended to be completed ***. Site personnel must also ***. It is critical, therefore, that site personnel review questionnaires for completeness at the time they are initially filled out, and that any missing answers are completed before the ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
7.3.2. ***
The *** for the assessment of *** that will be completed ***, and at *** after the ***.(22),(23)
Because this instrument is intended to be completed ***, it is important that *** on this questionnaire in any way. Should subjects ***. Because this questionnaire assesses the ***. Site personnel, therefore, must carefully review questionnaires for completeness before ***, and assure that questionnaires are properly completed.
The *** is being used ***. Answers to the questionnaire may reveal evidence of ***, including the ***. It is the responsibility of the investigator to evaluate ***. The evaluation by the Investigator will be guided by the ***. Investigators should document any such problems ***. It is expected that any randomized ***.
7.3.3. ***
The ***(24) is an ***. Each of the ***, and is answered on a yes/no basis. This assessment will be administered to all *** at *** in order to confirm the *** included in the treatment program. Subsequent *** evaluations will be done at *** after ***. All *** assessments must be administered by a trained staff member. If any test reveals ***, then test results must be confirmed by a physician investigator prior to ***.
7.4. ***
*** will be assessed using the ***. The ***will be assessed at ***, and ***, and measures ***. Effects of trial treatment on *** will be based on observed changes in the ***.
7.5. ***
*** assessments of *** and *** will be collected at ***. For the *** assessment, subjects will *** For the *** assessment, a ***
At the completion of study treatment (Visit 10), subjects will answer *** questions assessing ***. These questions are as follows:
· ***
· ***
· ***
7.6. Laboratory Tests
The following laboratory tests will be evaluated during this trial:
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Hematology tests, including hemoglobin, hematocrit, red blood cell count (RBC), total white blood cell count (WBC), WBC differential (neutrophils, lymphocytes, monocytes, eosinophils, basophiils) and platelet count, will be evaluated at screening and at Visit 10 (end of treatment or early exit);
· Fasting blood chemistries including: albumin, alkaline phosphatase, alanine transaminase (ALT), aspartate amino transferase (AST), blood urea nitrogen (BUN), serum calcium, serum chloride, serum sodium, carbon dioxide (CO2), creatinine (and estimated creatinine clearance), direct bilirubin, gamma-glutamyl transferase (GGT), glucose, lactate dehydrogenase (LDH), serum phosphorus, serum potassium, total bilirubin, total protein, and uric acid, will be evaluated at screening, Visit 4 (week 4), Visit 5 (week 8), Visit 7 (week 16), and Visit 10 (end of treatment or early exit);
· Lipid panel including total cholesterol, LDL cholesterol (LDL-C), HDL cholesterol (HDL-C), triglycerides, apolipoprotein A, apolipoprotein B-100, and Lp (a) will be evaluated at screening and Visit 10 (end of treatment or early exit). Visit 10 (end of treatment or early exit) results for apolipoprotein A, apolipoprotein B-100, and Lp (a) will be blinded to investigators and sponsor;
· Blood biomarkers including HgbA1c, C-reactive protein, adiponectin, retinol binding protein-4 (RBP-4), intercellular cell adhesion molecule (I-CAM), vascular cellular adhesion molecule (V-CAM), plasminogen activator inhibitor 1 (PAI-1) and fibrinogen, will be evaluated at screening and at Visit 10 (end of treatment or early exit). Visit 10 (end of treatment or early exit) results for all biomarkers with the exception of HgbA1c will be blinded to investigators and sponsor;
· Urine microalbumin, creatinine, and albumin/creatinine ratio will be evaluated at screening and at Visit 10 (end of treatment or early exit);
· Thyroid Stimulating Hormone (TSH) will be evaluated at screening;
· Serology testing for Hepatitis B surface antigen (HBsAg), Hepatitis C virus (HCV), and Human Immunodeficiency Virus (HIV) will be evaluated at screening;
· Urine drug screen for cannabinoids, cocaine, amphetamines, opiates, and phencyclidine will be evaluated at screening;
· Midstream UA with reflex microscopic evaluation will be evaluated at screening.
A certified central laboratory will be used for all of the above laboratory assessments.
In addition to the standard laboratory parameters described above, plasma samples will be obtained at Visit 7 (week 16) and Visit 10 (end of treatment or early exit) for the assessment of trough levels of phentermine and topiramate.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
For women of childbearing potential, urine pregnancy tests will be performed locally at each visit.
7.7. Physical Examination
A complete physical examination will be performed at week 0 (Visit 2) and week 28 (Visit 10 or early withdrawal). The physical examination will consist of an examination of the following systems: general appearance, skin, eyes, ears, nose, throat, neck (including thyroid), lymph nodes, chest, heart (including any auscultated cardiac murmurs), abdomen, extremities, and neurologic.
7.8. Electrocardiograms (ECG)
Twelve-lead electrocardiographic studies will be obtained at week 0 (Visit 2) and the end of treatment (Visit 10 or early withdrawal). Studies will be evaluated for clinically significant abnormalities that would prevent entry into the study, and for clinically relevant changes between screening and end of treatment. Parameters including R-R, QRS, QT, QTc intervals will also be recorded.
8. ADVERSE EVENT REPORTING
8.1. Adverse Events
Adverse events (AEs) are defined as any untoward medical occurrences in subjects administered the trial treatment, whether or not they have a causal relationship to the treatment. All observed or volunteered adverse events regardless of suspected causal relationship to the investigational product must be reported as described in the following sections.
Study investigators must collect sufficiently detailed information to describe adverse events, their character, onset, severity, relationship to study treatment, and resolution or other outcomes. Descriptions of neurological or psychological adverse events should be consistent with standard diagnostic criteria and terminology (such as DSM-IV) rather than general reports of symptoms. Whenever possible, description under a unifying diagnosis should be used in preference to a list of multiple signs or symptoms. Any adverse event judged by the investigator to be causally related to study treatment must be followed and documented by the investigator until the events or their sequelae resolve or stabilize at a level acceptable to the investigator, and VIVUS concurs with that assessment. Investigators must also determine whether any adverse event meets the specific criteria for classification as a Serious Adverse Event (SAE; see Section 8.2), which requires immediate notification to VIVUS or its designated representative.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
8.1.1. Severity Assessment
The investigator will assess the severity of all adverse events using the ***,***, or *** to describe the maximum intensity of each adverse event. For purposes of consistency, these intensity grades are defined as follows:
· ***;
· ***;
· ***.
Note the distinction between the severity and the seriousness of an adverse event. A *** is not necessarily a ***. For example, a headache may be *** but would not be classified as serious unless it met one of the criteria for ***.
8.1.2. Causality Assessment
Trial investigators are required to provide an assessment of causality, for all adverse events observed during this trial. This assessment will provide a determination of whether, in the investigators’ judgment, there exists a reasonable possibility that the investigational product caused or contributed to an adverse event. For this assessment, investigators must categorize the causality as either “related” or “not related.” For an adverse event to be considered “related” to the trial treatment, there should be evidence that the event follows a reasonable temporal sequence from the administration of trial treatment, or that the event follows a known response pattern to the drug. Causality would be further confirmed by improvement in an adverse event upon stopping the trial treatment, and reappearance of the event upon rechallenge.
8.1.3. Abnormal Test Findings
The criteria for determining whether an abnormal objective test finding should be reported as an adverse event are as follows:
· Test result is associated with accompanying symptoms, and/or
· Test result requires additional diagnostic testing or medical/surgical intervention, and/or
· Test result leads to a change in trial dosing or discontinuation from the trial, significant additional concomitant drug treatment, or other therapy, and/or
· Test result is considered by the investigator or sponsor to represent a clinically significant finding.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Merely repeating an abnormal test, in the absence of any of the above conditions, does not constitute an adverse event. Any abnormal test result that is determined to be an error does not require reporting as an adverse event.
8.2. Serious Adverse Events
As defined in the Code of Federal Regulations (21 CFR 312.32), a serious adverse event or serious adverse drug reaction is any untoward medical occurrence at any dose that:
· Results in death;
· Is life-threatening (immediate risk of death);
· Requires inpatient hospitalization or prolongation of existing hospitalization;
· Results in persistent or significant disability/incapacity;
· Results in congenital anomaly/birth defect.
Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered serious adverse drug experiences when, based on appropriate medical judgment, they may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes listed above. Adverse events that, in the investigator’s judgment, significantly jeopardized trial subjects or required medical or surgical intervention in order to prevent any of the outcomes listed above, should, therefore, be reported as serious adverse events.
8.2.1. Definition of Hospitalization
Adverse events reported from clinical trials that result in hospitalization or prolong an existing hospitalization are considered serious. Any initial admission (even if less than 24 hours) to a healthcare facility meets these criteria.
Outpatient ambulatory surgical procedures (same day surgeries) and routine emergency room treatment do not qualify as hospitalizations. Additionally, hospitalization in the absence of a precipitating, clinical adverse event is not in itself a serious adverse event. Examples include, but are not limited to:
· Admission for treatment of a preexisting condition not associated with the development of a new adverse event or with a worsening of the preexisting condition (eg, for work-up of persistent pre-treatment lab abnormality);
· Administrative admission (eg, for yearly physical exam);
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Optional admission not associated with a precipitating clinical adverse event (eg, for elective cosmetic surgery);
· Pre-planned treatments or surgical procedures should be noted in the baseline documentation for the entire protocol and/or for the individual subject.
Diagnostic and therapeutic non-invasive and invasive procedures, such as surgery, should not be reported as adverse events. However, the medical condition for which the procedure was performed should be reported if it meets the definition of an adverse event. For example, an acute appendicitis that begins during the adverse event reporting period should be reported as the adverse event, and the resulting appendectomy should be recorded as treatment of the adverse event.
8.3. Eliciting Adverse Event Information
The investigator is to report all directly observed adverse events and all adverse events spontaneously reported by the trial subject. In addition, trial subjects should be ***
Certain adverse events require prompt and specific action by the investigator in any clinical trial. The following sections describe additional requirements to ensure ***.
8.3.1. Eye Pain
At each visit, subjects will be queried regarding eye symptoms, *** Subject responses will be recorded as adverse events, where appropriate. If any subject reports eye pain and/or ***, the subject should be referred to ***. Treatment with study drug should be discontinued until the ***.
8.3.2. ***
All subjects will be screened for the presence and *** at *** and subsequently monitored at *** after the *** using a validated survey instrument *** designed for assessment of *** in a primary care setting. The *** is a *** module based directly on the diagnostic criteria for ***.
*** will also be assessed at *** and *** following the *** using the ***. Should this additional assessment indicate the presence ***. Any such event must be ***. Subjects must be ***.
Any *** must be ***.
8.4. Reporting Period
The reporting period for adverse events begins when the subject provides written informed consent and extends until *** after the last dose of the investigational product is administered, or until the subject is discontinued from the study, whichever is later. All adverse events that occur
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
during this period and are known to the investigator must be reported according to the requirements outlined in Section 8.5.
8.5. Reporting Requirements
All adverse events will be reported on the adverse event page(s) of the CRF. In addition, serious adverse events must also be reported on a separate Serious Adverse Event form. Where the same data are collected, the forms must be completed in a consistent manner. For example, the same adverse event term should be used on both forms. Adverse events should be reported using concise medical terminology on the CRFs as well as on the form for collection of serious adverse event information.
8.5.1. Serious Adverse Event Reporting Requirements
If a serious adverse event occurs, VIVUS is to be notified within 1 business day of awareness of the event by the investigator. In particular, if the serious adverse event is fatal or life-threatening, notification to VIVUS must be made immediately, irrespective of the extent of available adverse event information. This timeframe also applies to additional new information (follow-up) on previously forwarded serious adverse event reports.
***
For all serious adverse events, the investigator is obligated to pursue and provide information to VIVUS in accordance with the timeframes for reporting specified above. In addition, an investigator may be requested by VIVUS to obtain specific additional follow-up information in an expedited fashion. This information may be more detailed than that captured on the adverse event case report form. In general, this will include a description of the adverse event in sufficient detail to allow for a complete medical assessment of the case and independent determination of possible causality. Information on other possible causes of the event, such as concomitant medications and illnesses must be provided. In the case of a subject death, a summary of available autopsy findings must be submitted as soon as possible to VIVUS or its designated representative.
8.5.2. Non-Serious Adverse Event Reporting Requirements
Non-serious adverse events are to be reported on the adverse event CRFs, which are to be submitted to VIVUS.
8.5.3. ***
If any trial subject becomes or is found to be *** while receiving the investigational product, the investigator must discontinue trial treatment and submit this information to VIVUS on an ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
The investigator will follow the *** and then notify VIVUS of the outcome. The investigator will provide this information as a follow up to the ***.
For reported ***. The status of an ***.
If *** meet the criteria for classification as serious adverse events ***, the investigator should follow the procedures for reporting serious adverse events. Similarly, any *** that are considered to be adverse events should be reported as such on the appropriate CRF, however, *** need not be reported as an adverse event if there is no associated adverse outcome.
For reporting purposes, *** should be reported as serious adverse events, but because the ***.
9. DATA ANALYSIS/STATISTICAL METHODS
9.1. Sample Size Determination
To demonstrate the efficacy of a combination therapy, the combination (phentermine and topiramate) must be superior to both of the individual components that comprise the combination and placebo, at ***. The projected sample size of this trial is determined by the anticipated mean difference between the *** and the ***. In a previously conducted VIVUS *** trial, *** had a ***, compared to ***. In this trial, the effect of the *** is roughly equal to ***. Assuming the weight reduction for ***, and the weight reduction for ***, then the estimated effect of *** would be ***. Similarly, the estimated effect for *** would be ***. The effect of the *** is estimated as ***. Therefore, the estimated sample size is based on detecting a mean difference of *** between the ***. The pooled standard deviation for percent change in body weight from this *** trial was approximately ***. With 100 subjects in each of the *** in this trial, the trial should have more than *** power to demonstrate that the *** is an effective combination.
9.2. Efficacy Analysis
9.2.1. Analysis of Primary Endpoint
The primary calculated endpoints for this trial are based on the percent weight loss at week 28, and the percentage of subjects with at least 5% weight loss at week 28. The percent weight loss at week 28 will be calculated as ***.
The primary subject population is the Intent to Treat (ITT) population that consists of all subjects who are randomized, receive at least 1 dose of trial treatment, and have at least 1 post-dosing trial assessment. For subjects who discontinue treatment prior to trial completion, every attempt will be made to have them return to the clinic at week 28 for final assessments, regardless of when they discontinued trial treatment. For subjects who ***.
The primary objective of the statistical test is to confirm that at least one of the proposed phentermine/topiramate combinations evaluated in this trial is an effective combination therapy
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
for weight reduction. To demonstrate that a combination therapy is effective, it is necessary to show that a *** versus each *** comprising the ***, and *** versus ***, are all significant at the *** using ***. If any one of the *** does not reach the *** level for either of the co-primary endpoints, then the efficacy of that combination cannot be confirmed. A step-down multiple testing procedure will be used to control the overall type I error for this trial. The hypothesis that the *** is an effective combination therapy will be tested first. Once the efficacy of this *** is confirmed, then the efficacy of the *** will be tested using the same criteria. Comparisons between treatments for the endpoint of percent weight loss will be assessed using a *** model with factors of ***. Comparisons between treatment of the percentage of subjects with at least 5% weight loss at week 28 will be evaluated by ***.
Once the efficacy of both the *** has been confirmed, the difference in mean weight reduction between the *** therapies will be estimated. *** percent confidence intervals for the difference in mean percent body weight reduction between the two combination therapies will be derived.
9.2.2. Analysis of Secondary Endpoints
Secondary efficacy endpoints that are based on continuous data, including changes from baseline to week 28 in waist circumference and ***, will be evaluated in a manner similar to the primary endpoint. Categorical variables, including the proportion of subjects achieving 10% reductions in body weight at week 28 will be evaluated by ***. Analyses of secondary endpoints will also be based on the ITT population, and a step down strategy analogous to that used for the primary endpoint will be implemented to protect the overall alpha levels for these comparisons.
9.3. Analysis of Other Endpoints
Additional efficacy endpoints evaluated during this trial include *** of *** and ***, changes in primary and secondary outcomes over monthly intervals during the study, and Framingham 10-year risk assessments. The methodology for these comparisons will be similar to that used for primary and secondary endpoints.
*** will be obtained during this trial using a multiple trough sampling scheme, where samples are obtained from each subject at weeks 16 and 28 (or early exit). These data will be combined for analyses with data from other phase 3 trials that will utilize similar sampling schemes. *** will characterize *** for both drugs. The effects of various covariates including (but not limited to) ***, gender, race, *** and age will be evaluated.
9.4. Safety Analysis
Safety analyses will include tabular summaries of adverse event frequency, ***. Changes from baseline in *** evaluating *** will be summarized by treatment group using point estimates and *** confidence intervals. Since the proposed indication for the study medication is weight loss, summaries of total ***, which assesses ***, will also be presented to ensure that overall effects
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
on *** are not driven exclusively by the study medication’s known pharmacologic effect on appetite. Descriptive summaries of individual questionnaire items will also be presented.
9.5. Interim Analysis
No interim analyses are planned for this trial.
9.6. ***
***
10. QUALITY CONTROL AND QUALITY ASSURANCE
During trial conduct, VIVUS or its agent will conduct periodic monitoring visits to ensure that the protocol and GCPs are being followed. The monitors may review source documents to confirm that the data recorded on CRFs are accurate. The investigator and institution will allow VIVUS monitors or its agents and appropriate regulatory authorities direct access to all appropriate source documents to perform this verification.
The trial site and trial-related documents may be subject to review by the institutional review board (IRB)/independent ethics committee (IEC), and/or to quality assurance audits performed by VIVUS or its agents, and/or to inspection by appropriate regulatory authorities.
It is important that the investigator(s) and their relevant personnel are available during the monitoring visits and possible audits or inspections and that sufficient time is devoted to the process by the investigator and site personnel.
11. DATA HANDLING AND RECORD KEEPING
11.1. Case Report Forms / Electronic Data Record
As used in this protocol, the term case report form (CRF) should be understood to refer to either a paper form or an electronic data record or both, depending on the data collection method used in this trial.
A CRF is required and should be completed for each included subject. The completed original CRFs are the sole property of VIVUS and should not be made available in any form to third parties, except for authorized representatives of VIVUS or appropriate regulatory authorities, without written permission from VIVUS.
It is the investigator’s responsibility to ensure completion and to review and approve all CRFs. CRFs must be signed by the investigator or by an authorized staff member. These signatures serve to attest that the information contained on the CRFs is complete and accurate. At all times, the investigator has final personal responsibility for the accuracy and authenticity of all clinical and laboratory data entered on the CRFs. Subject source documents are the physician’s subject
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
records maintained at the trial site. In most cases, the source documents will be the hospital’s or the physician’s chart. In cases where the source documents are the hospital or the physician’s chart, the information collected on the CRFs must match those charts.
In some cases, the CRF may also serve as the source document. In these cases, VIVUS and the investigator must prospectively document which items will be recorded in the source documents and for which items the CRF will stand as the source document.
11.2. Record Retention
To enable evaluations and/or audits from regulatory authorities or VIVUS, the investigator agrees to keep records, including the identity of all participating subjects (sufficient information to link records, eg, CRFs and hospital records), all original signed informed consent forms, copies of all CRFs, serious adverse event forms, source documents, and detailed records of treatment disposition. The records should be retained by the investigator according to ICH, local regulations, or as specified in the Clinical Study Agreement, whichever is longer.
If the investigator relocates, retires, or for any reason withdraws from the trial, VIVUS should be prospectively notified. The trial records must be transferred to an acceptable designee, such as another investigator, another institution, or to VIVUS. The investigator must obtain written permission from VIVUS before disposing of any records, even if retention requirements have been met.
12. ETHICS
12.1. Institutional Review Board (IRB)/Independent Ethics Committee (IEC)
Regulations require that an IRB/IEC oversee all investigational drug trials. This board or committee, the makeup of which must conform to local and regional regulations, will approve all aspects of the study, including the protocol, advertising, and the form used to obtain written informed consent from trial subjects, prior to initiation of the trial. It is the responsibility of the investigator to have prospective approval of the trial protocol, protocol amendments, informed consent forms, and other relevant documents, eg, advertisements, if applicable, from the IRB/IEC. All correspondence with the IRB/IEC should be retained in the Investigator File. Copies of IRB/IEC approvals should be forwarded to VIVUS. All correspondence with the IRB/IEC should be retained in the Investigator File and copies forwarded to VIVUS, Inc.
All amendments to the protocol must be reviewed and approved by VIVUS and the IRB/IEC prior to implementation. The only circumstance in which an amendment may be initiated prior to IRB/IEC approval is where the change is necessary to eliminate apparent immediate hazards to the subjects. In that event, the investigator must notify the IRB/IEC and VIVUS in writing within 5 working days after the implementation.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
The investigator is responsible for obtaining annual (at a minimum) IRB/IEC renewal for the duration of the trial. The investigator is also responsible for keeping the IRB/IEC advised of the progress of the trial, and of any changes made to the protocol. Copies of these reports and documentation of all IRB/IEC action (extension or otherwise) must be forwarded to VIVUS.
12.2. Ethical Conduct of the Trial
The trial will be performed in accordance with the protocol, International Conference on Harmonization Good Clinical Practice guidelines, and applicable local regulatory requirements and laws.
12.3. Subject Information and Consent
The informed consent form and any changes to the informed consent form made during the course of the trial must be agreed to by VIVUS, Inc. and the IRB/IEC prior to its use and must be in compliance with all ICH-GCP, local regulatory requirements and legal requirements.
The investigator must ensure that each trial subject is fully informed about the nature and objectives of the trial and possible risks associated with participation and must ensure that the subject has been informed of his/her rights to privacy. The investigator will obtain written informed consent from each subject before any trial-specific activity is performed and should document in the source documentation that consent was obtained prior to enrollment in the trial. The original signed copy of the informed consent form must be maintained by the investigator and is subject to inspection by a representative of VIVUS, Inc., their representatives, auditors, the IRB/IEC and/or regulatory agencies. A copy of the signed informed consent form will be given to the subject.
12.4. Disclosure of Data
Data generated by this trial must be available for inspection by the U.S. Food and Drug Administration (FDA), by the sponsor or a designate acting on behalf of the sponsor, by applicable foreign health authorities, and by the IRB or EC as appropriate. At a subject’s request, medical information may be given to their personal physician or other appropriate medical personnel responsible for their welfare.
Subject medical information obtained during the course of this trial is confidential and disclosure to third parties other than those noted above is prohibited.
13. Regulatory Correspondence
The trial site and trial-related documents may be subject to review by the institutional review board IRB/IEC, and/or to quality assurance audits performed by VIVUS, Inc., and/or to inspection by the FDA and/or applicable foreign health authorities. The investigator will notify VIVUS, Inc. within *** working days following any FDA or other regulatory agency contact
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
with the investigative site regarding this study. The investigator will provide VIVUS, Inc. with copies of all correspondence with the FDA or other regulatory agency which may affect the review of the current study (e.g., Form 483, Inspection Observations) or their qualification as an investigator in studies conducted by VIVUS, Inc. (e.g., warning letters).
14. Definition of end of trial (If Applicable)
The end of this trial is defined as the time when the last subject completes their last trial visit.
Additionally, data and materials that are required by the Sponsor before any trial site’s activity can be considered completed include:
· All completed Case Report Forms, appropriately signed by the investigator;
· All laboratory findings, clinical data, and special test results collected during the trial period;
· Completed drug accountability and investigational materials return records;
· Statement of outcome for any serious adverse events reported during the study;
· Copy of notification to IRB/IEC indicating study completion.
15. SPONSOR DISCONTINUATION CRITERIA
Premature termination of this clinical trial may occur because of a regulatory authority decision, change in opinion of the IRB/IEC, drug safety problems, or at the discretion of VIVUS. In addition, VIVUS retains the right to discontinue development of VI-0521 at any time.
If a trial is prematurely terminated or discontinued, VIVUS will promptly notify the investigator. After notification, the investigator must contact all participating subjects within 15 days. As directed by VIVUS, all trial materials must be collected and all CRFs completed to the greatest extent possible.
16. PUBLICATION OF TRIAL RESULTS
All information and data, including the terms of this protocol, and all data, clinical results, and research conducted hereunder concerning VIVUS, Inc.’s products and operations including VIVUS, Inc. patent applications, formulas, manufacturing processes, basic scientific data, and formulation information that has been supplied by VIVUS, Inc. and not previously published are considered confidential by VIVUS, Inc. and will remain the sole property of VIVUS, Inc. The investigator understands and agrees that said proprietary and/or confidential information disclosed to or produced by him/her thereunder is highly valuable to VIVUS, Inc. and will be used exclusively by the investigator in accomplishing this study and will not use it for any other
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
purposes without VIVUS, Inc’s prior written consent. The investigator agrees that he/she will not use any such proprietary and/or confidential information for any other purpose. The investigator also understands and agrees that such disclosure will not be deemed to grant to the investigator a license for use of said proprietary and/or confidential information, except as expressly provided herein.
It is understood by the investigator that the information developed in the clinical study will be used by VIVUS, Inc. in connection with the development of this product. This information, therefore, may be disclosed and used solely by VIVUS, Inc. as required to such third parties and agencies as VIVUS, Inc., in its sole discretion, warrants. In order to allow for the use of the information derived from the clinical studies, it is understood that there is an obligation to provide to VIVUS, Inc. complete test results and all data developed in this study. The investigator agrees to promptly answer all inquires from VIVUS, Inc. regarding completion, legibility or accuracy of trial data in the case report.
VIVUS, Inc. recognizes the value of disseminating research results and expects that publication of all results from this study will be undertaken by a collaborative group of study investigators who made significant contributions to the study design, the treatment of study subjects, and evaluation of study data. However, after 1) submission of the multicenter results for publication, 2) notification ***.
Investigators shall furnish the *** with a written copy of any proposed publication or other disclosure of study results (including disclosures at research seminars, lectures and professional meetings) *** prior to submission for publication or disclosure so that *** may have a reasonable opportunity to protect its proprietary rights to information, inventions, or products developed under this study and to insure that reported data are factually correct. Upon the ***, the investigator shall not publish or disclose information related to this study. Further, if the *** believes that such publication or disclosure contains confidential information, the investigator agrees to remove such confidential information from the proposed publication or disclosure.
VIVUS, Inc. agrees that before it publishes any results of this study in a refereed journal, it will provide the investigator, for review, a prepublication manuscript *** prior to the submission of the manuscript to the publisher.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
17. REFERENCES
(1) Adipex-P® (Phentermine Hydrochloride USP, 37.5 mg) Package Insert (2005). Gate Pharmaceuticals, Sellersville, PA.
(2) Topamax® (topiramate) Tablets and Topamax® (topiramate capsules) Sprinkle Capsules Package Insert (June 2003). Ortho-▇▇▇▇▇▇ Pharmaceutical, Inc., ▇▇▇▇▇▇▇, ▇▇ ▇▇▇▇▇.
(3) ▇▇▇▇▇▇ K, ▇▇▇▇▇▇▇ M, ▇▇▇▇▇ C, ▇▇▇▇▇▇▇▇ C. Prevalence and trends in obesity among US adults, 1999-2000. JAMA 2002;288:1723-27.
(4) ▇▇▇▇▇▇▇ P, ▇▇▇▇▇ T, ▇▇▇▇ G, et al. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss: An update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Commission of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 2006;113:898-918.
(5) ▇▇▇▇▇ C, ▇▇▇▇▇▇▇ M, ▇▇▇▇▇▇ L, et al. Prevalence of overweight and obesity in the ▇▇▇▇▇▇ ▇▇▇▇▇▇, ▇▇▇▇-▇▇▇▇. JAMA 2006;295:1549-55.
(6) Must A, Spadano J, ▇▇▇▇▇▇▇ E, et al. The disease burden associated with overweight and obesity. JAMA 1999:282;1523-29.
(7) Katzmarzyk P, ▇▇▇▇▇▇▇ I, Ardern C. Physical inactivity, excess adiposity and premature mortality. Obes Res 2003;4:257-90.
(8) ▇▇▇▇▇ K, ▇▇▇▇▇▇▇▇▇ A, ▇▇▇▇▇ T, et al. Overweight, obesity, and mortality in a large prospective cohort of persons 50 to 71 years old. NEJM 2006;355:763-78.
(9) ▇▇▇▇▇▇▇▇▇ D. Beneficial health effects of modest weight loss. Int J Obes 1992;6:297-415.
(10) Pasanisi F, Contaido F, ▇▇▇▇▇▇▇▇ G, ▇▇▇▇▇▇▇ M. Benefits of sustained moderate weight loss in obesity. Nutr Metab Cardiovasc Dis 2001;11:401-6.
(11) Douketis J, ▇▇▇▇▇ C, Thabane L, ▇▇▇▇▇▇▇▇▇▇ D. Systematic review of long-term weight loss studies in obese adults: Clinical significance and applicability to clinical practice. Int J Obes 2005;10:1153-67.
(12) ▇▇▇▇▇▇ R, Majumdar S. Drug treatments for obesity: Orlistat, sibutramine, and rimonabant. Lancet 2007;369:71-7.
(13) Montague C, Farooqi I, ▇▇▇▇▇▇▇▇▇ J, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997;387:903-8.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
(14) Heymsfield S, ▇▇▇▇▇▇▇▇▇ A, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: A randomized, controlled, dose-escalation trial. JAMA 1999;282:1568-75.
(15) ▇▇▇-▇▇▇▇▇▇▇▇ E, ▇▇▇▇▇▇▇ M, ▇▇▇▇▇▇▇▇ E, et al. Predictors of weight loss in adults with topiramate-treated epilepsy. Obes Res 2003;11:556-62.
(16) ▇▇▇▇ G, ▇▇▇▇▇▇▇▇▇ P, ▇▇▇▇▇ S, et al. A 6-month randomized, placebo-controlled, dose-ranging trial of topiramate for weight loss in obesity. Obes Res 2003;11:722-33.
(17) ▇▇▇▇▇▇▇ J, ▇▇▇▇▇▇▇ L, ▇▇▇▇▇▇▇▇ A, et al. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of topiramate in the treatment of obese subjects. Int J Obes Relat Metab Disord 2004;28:1399-410.
(18) Clinical Study Report: A Phase II, 24-Week, Randomized, Double-Blind, Placebo-Controlled, Single-Center Study to Examine Safety, Tolerability and Efficacy of Topiramate and Phentermine Combination Therapy for Weight Loss in Healthy Obese Subjects. Data on file. VIVUS, Inc., Mountain View, CA.
(19) ▇▇▇▇▇▇ P, ▇’▇▇▇▇▇▇▇▇ R, ▇▇▇▇ D, et al. Prediction of Coronary Heart Disease Using Risk Factor Categories. Circulation 1998;97:1837-47.
(20) ***
(21) ***
(22) ***
(23) ***
(24) ***
(25) ***
(26) ***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 1: SCHEDULE OF STUDY ACTIVITIES
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
| ||||||||||||
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
|
|
|
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
*** |
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 2: ***
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 3: ***
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 4: PROTOCOL AMENDMENTS
Amendment Tracking
|
Protocol Title: |
A Phase III, Randomized, Double-Blind, parallel-design study comparing multiple doses of VI-0521 to Placebo and their single-agent phentermine and topiramate constituents for the Treatment of Obesity in Adults |
|
|
|
|
Protocol Number: |
OB-301 |
Indicate any amendments to this protocol by writing the Amendment Number and the Date of the Amendment in the space below. This page will be used to track the original protocol and its amendments. This is the original protocol if no amendment number is listed below.
Amendment Number 3
Date of Amendment: ***
(19 items)
Rationale: This protocol amendment is being implemented at the request of the FDA to incorporate assessments of *** and *** using the ***, and assessments of *** using the *** in all study subjects at ***. Previously, follow-up *** evaluations were done only if other *** assessments revealed a ***. This change is not anticipated to have any impact on safety of study subjects.
|
Section and/or |
|
Protocol |
|
Amendment 3, *** |
|
|
|
|
|
|
|
Protocol Synopsis: |
*** |
Change: “Adverse event assessments will include direct questions related to eye pain and *** at each visit. In addition, the presence of *** will be evaluated at baseline using the ***. Follow up assessments using the *** will be done if adverse events potentially related to ***, or ***.”
To: “Adverse event assessments will include direct questions related to eye pain and *** will be assessed at each visit using the ***.” | ||
|
|
|
| ||
|
Rationale |
*** |
Change: “VI-0521 is an exploratory weight loss therapy that is a new combination of two currently approved drugs, phentermine and topiramate. As such, VI-0521 represents a potential advance in the medical treatment of obesity since the two agents comprising this combination product affect weight loss through different mechanisms.”
To: “VI-0521 is an investigational weight loss therapy that is a combination of two currently approved drugs, phentermine and topiramate. As such, VI-0521 represents a potential advance in the medical treatment of obesity since the two | ||
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
agents comprising this combination product effect weight loss through different mechanisms.” |
|
|
|
|
|
Trial Design |
*** |
Change: “Safety evaluations will include summaries of adverse events, ***.”
To: “Safety evaluations will include summaries of adverse events, ***.” |
|
|
|
|
|
Drug Storage and Drug Accountability |
*** |
Change: “No investigational drug, used or unused, may be discarded. All used and unused drug must be returned to the sponsor upon completion of the study.”
To: “No investigational drug, used or unused, may be discarded. All used and unused drug must be returned to the sponsor or designated representative upon completion of the study.” |
|
|
|
|
|
Schedule of Visits: Visit 2 |
*** |
Change: “Assess adverse events (including eye pain and ***), and changes in concomitant medications;”
To: “Assess adverse events (including eye pain), and changes in concomitant medications;” |
|
|
|
|
|
Schedule of Visits: Visit 3 |
*** |
Added: “Administer *** and ***;”
Change: “Assess adverse events (including eye pain and ***), and changes in concomitant medications;”
To: “Assess adverse events (including eye pain), and changes in concomitant medications;” |
|
|
|
|
|
Schedule of Visits: Visit 4 |
*** |
Added: “Administer *** and ***;”
Change: “Assess adverse events (including eye pain and ***), and changes in concomitant medications;”
To: “Assess adverse events (including eye pain), and changes in concomitant medications;” |
|
|
|
|
|
Schedule of Visits: Visit 5 |
*** |
Added: “Administer *** and ***;” Change: “Assess adverse events (including eye pain and ***), and changes in concomitant medications;” To: “Assess adverse events (including eye pain), and changes in concomitant medications;” |
|
|
|
|
|
Schedule of Visits: Visit 6 |
*** |
Added: “Administer *** and ***;”
Change: “Assess adverse events (including eye pain and ***), and changes in concomitant medications;”
To: “Assess adverse events (including eye pain), and changes in concomitant medications;” |
|
|
|
|
|
Schedule of Visits: Visit 7 |
*** |
Change: “Administer ***;”
“Assess adverse events (including eye pain and ***), and changes in concomitant medications;”
To: “Administer *** and ***;”
“Assess adverse events (including eye pain), and changes in concomitant medications;” |
|
|
|
|
|
Schedule of Visits: Visit 8 |
*** |
Added: “Administer *** and ***;”
|
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
Change: “Assess adverse events (including eye pain and ***), and changes in concomitant medications;”
To: “Assess adverse events (including eye pain), and changes in concomitant medications;” |
|
|
|
|
|
Schedule of Visits: Visit 9 |
*** |
Added: “Administer *** and ***;”
Change: “Assess adverse events (including eye pain and ***), and changes in concomitant medications;”
To: “Assess adverse events (including eye pain), and changes in concomitant medications;” |
|
|
|
|
|
Schedule of Visits: Visit 10 |
*** |
Change: “Administer *** and ***;” “Assess adverse events (including eye pain and ***), and changes in concomitant medications;” To: “Administer ***, and ***;” “Assess adverse events (including eye pain), and changes in concomitant medications;”
|
|
|
|
|
|
*** |
*** |
Change: “The *** for the assessment of *** that will be completed ***.(22),(23)”
To: “The *** for the assessment of *** that will be completed ***, and at *** after the ***.(22),(23)”
Change: “The *** is being used ***. This questionnaire will be completed at ***, and ***.”
To: “The *** is being used ***.” |
|
|
|
|
|
*** |
*** |
Change: “Subsequent *** evaluations will be done only in *** who demonstrate a ***.” To: “Subsequent *** evaluations will be done at *** after ***. All *** assessments must be administered by a trained staff member. If any test reveals ***, then test results must be confirmed by a physician investigator prior to ***.” |
|
|
|
|
|
*** |
*** |
Change:” All subjects will be screened for the presence and *** at *** and subsequently at regular intervals throughout the study: *** and *** using a validated survey instrument *** designed for assessment of *** in a primary care setting.”
To: “All subjects will be screened for the presence and *** at *** and subsequently monitored at *** after the *** using a validated survey instrument *** designed for assessment of *** in a primary care setting.”
Change: “The *** is a *** module based directly on the diagnostic criteria for ***. At study visits where *** assessments are not included, subjects will be asked the following question to assess ***.”
To: “The *** is a *** module based directly on the diagnostic criteria for ***.
*** will also be assessed at *** and *** following the *** using the ***.”
Change: “Any *** must be ***. Investigators are encouraged to administer the *** on an ad-hoc basis as part of the clinical assessment of ***. These ad-hoc questionnaires will be maintained as source documentation, but will not be |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
analyzed as a separate ***.”
To: “Any *** must be ***.” |
|
|
|
|
|
Appendix 1: Schedule of Study Activities — *** |
*** |
Add: *** |
|
|
|
|
|
Appendix 1: Schedule of Study Activities — *** |
*** |
Add: *** |
|
|
|
|
|
Appendix 1: Schedule of Study Activities — *** |
*** |
Change: “***” To: “***” |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Protocol Title: A Phase III, Randomized, Double-Blind, parallel-design study comparing multiple doses of VI-0521 to Placebo and their single-agent phentermine and topiramate constituents for the Treatment of Obesity in Adults
Protocol Number: OB-301
Indicate any amendments to this protocol by writing the Amendment Number and the Date of the Amendment in the space below. This page will be used to track the original protocol and its amendments. This is the original protocol if no amendment number is listed below.
Amendment Number 2
Date of Amendment: ***
(14 items)
Rationale: This protocol amendment is being implemented in response to comments from FDA and investigational sites to designate the percent weight loss from baseline to week 28 and the percentage of subjects achieving at least 5% weight loss as co-primary endpoints, to provide an additional *** at ***, and to specify that ***. Additionally miscellaneous inconsistencies and typographical errors have been addressed. Other than the potentially positive effects of ***, these changes are not anticipated to have any impact on subject safety.
|
Section and/or |
|
Protocol |
|
Amendment 2, *** |
|
|
|
|
|
|
|
Header |
|
*** |
|
Changed: “ Amendment 1: ***” To: “Amendment 2: ***” |
|
|
|
|
|
|
|
Protocol Synopsis: Efficacy Endpoints |
|
*** |
|
Change: “The primary study endpoints will be based on the percent weight loss at week 28, calculated as ***.”
To: “The primary study endpoints will be based on the percent weight loss at week 28, calculated as ***, and the percentage of subjects achieving at least 5% weight loss at week 28.” |
|
|
|
|
|
|
|
Protocol Synopsis: Efficacy Endpoints |
|
*** |
|
Change: “Secondary efficacy endpoints will include the proportion of subjects achieving 5% and 10% weight loss,”
To: “Secondary efficacy endpoints will include the proportion of subjects achieving 10% weight loss,” |
|
|
|
|
|
|
|
Protocol Synopsis: Statistical Methods |
|
*** |
|
Change: “If each of these *** comparisons is significant at the ***, then this combination will be considered to have met requirements,”
To: “If each of these *** comparisons is significant at the *** for both of the co-primary endpoints, then this combination will be considered to have met requirements,” |
|
|
|
|
|
|
|
Trial Design |
|
*** |
|
Change: “The primary efficacy endpoints for this trial will be based on the |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
percent weight loss at week 28, calculated as ***. Secondary efficacy endpoints will include the percentage of subjects losing 5% and 10% of their body weight,”
To: “ The primary efficacy endpoints for this trial will be based on the percent weight loss at week 28, calculated as ***, and the percentage of subjects achieving at least 5% weight loss at week 28. Secondary efficacy endpoints will include the percentage of subjects losing 10% of their body weight,” |
|
|
|
|
|
|
|
Schedule of Visits: Visit 4 |
|
*** |
|
Change: “Administer *** for *** and ***;”
To: “Administer *** for *** and ***, and perform *** (see Section 7.4);” |
|
|
|
|
|
|
|
Schedule of Visits: Visit 10 |
|
*** |
|
Change: “Collect blood sample for laboratory tests (all subjects),”
To: “Collect blood and urine samples for laboratory tests (all subjects),” |
|
|
|
|
|
|
|
Schedule of Visits: Visit 10 |
|
*** |
|
Add: “Ask End of Treatment Questions (see Section 7.5);”
Change: “Discontinue subject’s trial participation; if appropriate enroll subject into open label treatment protocol.”
To; “Discontinue subject’s trial participation.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “The *** measures ***”
To: “The *** will be assessed at ***, and ***, and measures ***,” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “*** assessments of *** and *** will be collected at selected visits at ***.”
To: “*** assessments of *** and *** will be collected at ***.” |
|
|
|
|
|
|
|
Analysis of Primary Endpoint |
|
*** |
|
Change: “The primary calculated endpoint for this trial is based on the percent weight loss at week 28. This outcome will be calculated as ***.”
To: “The primary calculated endpoints for this trial are based on the percent weight loss at week 28, and the percentage of subjects with at least 5% weight loss at week 28. The percent weight loss at week 28 will be calculated as ***.” |
|
|
|
|
|
|
|
Analysis of Primary Endpoint |
|
*** |
|
Change: “If any one of the *** does not reach the *** level, then the efficacy of that combination cannot be confirmed. A step-down multiple testing procedure will be used to control the overall type I error for this trial. The hypothesis that the *** is an effective combination therapy will be tested first. Once the efficacy of this *** is confirmed, then the efficacy of the *** will be tested using the same criteria. Comparisons between treatments will be assessed using a *** model with factors of ***.”
To: “If any one of the *** does not reach the *** level for either of the co-primary endpoints, then the efficacy of that combination cannot be confirmed. A step-down multiple testing procedure will be used to control the overall type I error for this trial. The hypothesis that the *** is an effective combination therapy will be tested first. Once the efficacy of this *** is confirmed, then the |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
efficacy of the *** will be tested using the same criteria. Comparisons between treatments for the endpoint of percent weight loss will be assessed using a *** model with factors of ***. Comparisons between treatment of the percentage of subjects with at least 5% weight loss at week 28 will be evaluated by ***.” |
|
|
|
|
|
|
|
Analysis of Secondary Endpoint |
|
*** |
|
Change: “Categorical variables, including the proportion of subjects achieving 5% and 10% reductions in body weight at week 28 will be evaluated by ***.”
To: “Categorical variables, including the proportion of subjects achieving 10% reductions in body weight at week 28 will be evaluated by ***.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “This trial will not involve a ***.”
To: “***.” |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Protocol Title: A Phase III, Randomized, Double-Blind, parallel-design study comparing multiple doses of VI-0521 to Placebo and their single-agent phentermine and topiramate constituents for the Treatment of Obesity in Adults
Protocol Number: OB-301
Indicate any amendments to this protocol by writing the Amendment Number and the Date of the Amendment in the space below. This page will be used to track the original protocol and its amendments. This is the original protocol if no amendment number is listed below.
Amendment Number 1
Date of Amendment: ***
(8 items)
|
Section and/or |
|
Protocol |
|
Amendment 1, *** |
|
|
|
|
|
|
|
Appendix 4 |
|
*** |
|
Added: Appendix 4: Protocol Amendment Section |
|
|
|
|
|
|
|
List of Abbreviations |
|
*** |
|
Add: “I-CAM: Intercellular Cell Adhesion Molecule”, “PAI-1: Platelet Activator Inhibitor 1”, “RBP-4: Retinol Binding Protein 4”, and “V-CAM: Vascular Cell Adhesion Molecule” |
|
|
|
|
|
|
|
Inclusion Criteria |
|
*** |
|
Change: “If females of child-bearing potential, be using adequate contraception, defined as double barrier methods, stable hormonal contraception plus single barrier, or tubal ligation. Females are considered to be of childbearing potential unless they are at least 55 years of age with spontaneous amenorrhea for at least 12 months, have a documented FSH value of 40 IU/L or greater, or have had a bilateral hysterectomy or oophorectomy;
To: “If females of child-bearing potential, be using adequate contraception, defined as double barrier methods, stable hormonal contraception plus single barrier, or tubal ligation. Females are considered to be of childbearing potential unless they are at least 55 years of age with spontaneous amenorrhea for at least 12 months or have a documented FSH value of 40 IU/L or greater, or have had a hysterectomy or bilateral oophorectomy;” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “Should *** would cause subjects to consider ***, investigators may, at their discretion, ***. Such events may or may not be related to trial treatment.
If dosing has been ***, a new titration kit should be ordered through *** to ***.”
To: “Should *** would cause subjects to consider ***, investigators may, at their discretion, ***. *** are possible with agreement from the medical monitor. Such events may or may not be related to trial treatment. |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #01
Appendix 2 — Scope of Work
VIVUS, Inc.
Qnexa OB-301
PROJECT OVERVIEW
▇▇-▇▇▇
▇▇▇ ▇▇-▇▇▇ trial is a phase III, randomized, double-blind, parallel design study comparing multiple doses of VI-0521 to placebo and their single-agent phentermine and topiramate constituents for the treatment of obesity in adults.
PROJECT TEAM
Study Management
The overall management of the study will be the responsibility of the Senior Clinical Trial Manager (CTM). The Senior CTM will oversee and coordinate the management of the study as well as oversee the study specific CTM. This oversight will ensure consistency and allow VIVUS Study Management to have one primary contact for the Qnexa program. The Medpace CTM assigned to OB-301 will work closely with the VIVUS Study Manager, Medpace Medical Expert, and VIVUS Clinical Leader to address protocol questions and interpretations while maintaining close oversight of study-related processes and documents. The OB-301 CTM will supervise all Clinical Research Associates (CRAs) and Project Coordinator assigned to the project.
The Project Coordinator will be responsible for day-to-day study management functions, including the generation of status reports, organization of supplies, generation and compilation of newsletters, and input of all study information into the ClinTrak® Study Management System, a web-based, proprietary research management system designed by Medpace. The Project Coordinator will organize teleconferences and team meetings, including the compilation of agendas and meeting minutes.
The Study Start-Up Manager and Study Start-Up Coordinators will work closely with the CTM and Project Coordinator to ensure sites become active in the most time effective manner.
The Medpace Contracts Attorney will be responsible for the execution of Investigator contracts (upon VIVUS defined process). The Contracts Attorney will work closely with the Start-Up Manager and Medpace CTM to ensure contracts are executed in a timely manner.
The Medpace Medical Expert assigned to this project will work closely with the VIVUS Clinical Leader. The Medpace Medical Expert will assist with protocol design and medical interpretation of entry criteria and adverse events (AEs). The Medical Expert will also be involved in the training of CRAs and other staff members participating in the
|
CONFIDENTIAL |
|
September 7, 2007 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
project. The Medical Expert will review and approve the coding of concomitant medications, medical histories, AEs, and will provide the medical context for the statistical analysis and medical writing.
The Medical Expert will assist in the review of the protocol, train Medpace personnel internally as to the background of the study compound and design of the study, participate in the project teleconferences and meetings, work hand-in-hand with the OB-301 CTM, and have heavy involvement in the clinical study report. The Medical Expert’s role and decision making rights are dictated by VIVUS (e.g. inclusion/exclusion of patients, discussions with Investigators about withdrawing a patient, etc.). This decision making power often times reduces the oversight needed by the sponsor. For questions the OB-301 CTM is not comfortable answering, she will contact the Medical Expert for guidance. Obviously, VIVUS will be involved in study oversight based on pre-defined terms with the VIVUS Clinical Development Team. The Medpace Medical Expert is available 24 hours a day, 7 days a week via the Medpace Project Helpline.
Clinical Monitoring
Medpace operates in North America with a primarily centralized monitoring team of over 140 CRAs to promote greater standardization, cohesiveness, support, and stability. Each of the Medpace CRAs assigned to this project have monitoring experience and strong clinical backgrounds.
Clinical Safety
The Clinical Safety will be managed by VIVUS or its designee. VIVUS Clinical Leader to be involved with casualty assignment for all Serious Adverse Events (SAEs).
Data Management
A Data Manager will serve as the primary contact for the Data Management team. Data Coordinators will be involved in the day-to-day operations and report issues to the Data Manager. Data Entry Specialists and Database Programmers will also be utilized.
Biometrics
Key members of our Biometrics team include Biostatisticians and Statistical Analysts. The Biostatistician assigned to this project will develop the analysis plan and coordinate biometrics activities. The Lead Statistical Analyst will work closely with the Biostatistician to ensure a clear understanding of the analysis plan and communicate any programming issues that may arise.
Medical Writing
The Medical Writing team works in collaboration with the Medical Experts to prepare research reports meeting International Conference on Harmonisation (ICH) and Sponsor guidelines. All Medpace Medical Writers have extensive experience in regulatory
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
submission preparation. The Medical Writing team is actively involved throughout the conduct of the trial.
Team Organization Chart
PROJECT START-UP
Protocol
VIVUS will prepare the protocol. Medpace will review the protocol and provide comments before it is finalized.
Case Report Forms
Medpace will design the electronic case report forms (eCRFs) for the trial, including completion instructions, according to the final protocols and the Medpace template. VIVUS must review and approve the eCRFs before they are finalized.
Project Initiation
Prior to the study site initiation visits, a project kickoff meeting will be held at Medpace involving Medpace and VIVUS personnel to review the study protocol, eCRFs, and overall project coordination. Medpace project team members and VIVUS personnel will participate in this meeting.
Interactive Voice Response System
Medpace will provide a customized (study-specific) interactive voice response system (IVRS) to provide patient randomization, and drug management. The Medpace IVRS is a proprietary in-house developed system. The system provides both voice and web access and has been developed in conjunction with our web based Clintrak® system providing
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
seamless functionality throughout the conduct of the study. The VIVUS Team (no limit applied to number of team members) will have access to review reports within the IVRS. Medpace will perform the User Acceptance Testing (UAT) for each site.
The IVRS will include:
· Subject Screens/Screen Failures;
· Subject randomization;
· Patient visit tracking;
· Inventory management (site supply set-up, initial bulk supply, and resupply of one additional shipment per patient);
· Notifications of site shipments;
· Confirmation of receipt of shipments; and
· Customized reports.
VIVUS will review IVRS and approve system prior to finalization.
Study Medication Supply and Storage
VIVUS will be responsible for the supply, packaging, labeling, storage, and destruction of study medication. Distribution of the study medication will be tracked and initiated via the Medpace IVRS. Study medication accountability procedures will follow Medpace standard operating procedures (SOPs) and utilize a study medication accountability log that has been approved by VIVUS.
Recruitment Oversight Plan
The Medpace CTM, in collaboration with VIVUS, will develop a recruitment oversight plan. Medpace understands the importance of rapid recruitment and the necessity to keep patients in the trial until completion. Medpace will develop processes prior to study initiation to ensure recruitment is efficient and retention of patients in the study is maximized. The plan will include details on:
· Initial collection of essential documents;
· Patient recruitment (including tools, site-specific plans, contingencies, etc.); and
· Patient recruitment tracking reports.
The tools noted above include:
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Inclusion/exclusion cards;
· Patient emergency cards;
· Enrollment tracking forms;
· Laminated patient visit schedule;
· Pocket protocol; and
· Advertising tools (e.g., posters, table tents, language for newspaper/radio advertisements, etc.).
Medpace understands each site has a different experience and approach to patient recruitment. The Medpace CTM and Medpace CRAs will work with personnel from each site to help maximize their efforts. The project team maintains frequent contact during the active recruitment phase to assess each site’s activity and offer assistance when needed. Medpace acknowledges the importance of site recognition and offers various incentives throughout the recruitment period. Examples of site incentives are “Site of the Month” awards and weekly faxes displaying each sites’ activity (often included in the newsletter as well). The time associated with these types of incentives are inclusive of the budget. However, often times, monetary incentives are built into the Investigator’s grant to encourage rapid essential document collection and/or timely recruitment.
Patient retention is vital to the success of a trial. The Medpace CTM and the Medpace CRAs will work with the sites to understand the needs and motivations for patients to remain in the study. They will help educate the sites on the importance of “customer service” and “patient satisfaction” as elements that ensure continued patient participation. Examples of patient retention methods include quarterly patient newsletters, ideas for site customer service, and tokens of appreciation for patients.
Site Selection and Pre-study Visits
VIVUS, in conjunction with Medpace, will identify qualified Investigators. VIVUS and Medpace will also work together to provide and negotiate Confidentiality Disclosure Agreements (CDAs) as well as create and evaluate site questionnaires. In not knowing the number of pre-study visits Medpace will be required to conduct, a unit price for a pre-study visit has been provided in the budget.
Pre-study visits will be conducted consistent with Medpace SOPs. Medpace will provide a pre-study visit report to VIVUS within 10 business days of each visit.
These visits will include, but are not limited to:
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Determining whether or not the site has clinical staff of appropriate education, training, and experience to manage the study and have sufficient capacity to perform the required tasks;
· Determining whether or not the site has appropriate facilities to conduct the study;
· Determining there are no competing studies that will conflict with patient enrollment and that the site has sufficient patients and processes to enroll patients in the time identified for patient recruitment; and
· Determining whether or not the site has appropriate resources and procedures to maintain appropriate records according to FDA requirements.
Study Start-up Team
The efficient start-up of the *** study sites for OB-301 will have a significant effect on patient recruitment time. Medpace will utilize its Study Start-up team for additional support during the initial phase of the trials to expedite the overall study start-up for each site.
The Study Start-up team works directly with the CTM and the Project Coordinators. The team is comprised of a Study Start-up Manager and several experienced Study Start-up Coordinators. The team is responsible for many of the key start-up activities, including:
· Submission to the central IRB;
· Coordination and tracking of essential documents packages for each site;
· Investigator meeting presentations and binders; and
· Site tools.
Central Laboratory Selection
Medpace Reference Laboratories (MRL) will be utilized for processing the clinical laboratory samples. MRL is committed to providing comprehensive laboratory services of the highest quality to the pharmaceutical and biotechnology industries.
Investigators’ Meeting
An Investigators’ Meeting will be held for the OB-301 study. VIVUS will arrange the meeting (including contracting with a third-party vendor) and Medpace will prepare the meeting materials, including preparation and distribution of binders. The Medpace OB-301 Team will attend the meeting. VIVUS will open the meeting and Medpace will present on the topics delegated by VIVUS. The meeting minutes will be prepared by Medpace, reviewed and approved by VIVUS, and distributed to the study sites by
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Medpace. The preparation of the meeting minutes is optional; however, is included in the budget. Medpace assumes the Investigators’ Meeting will serve as the initiation visit for the Investigators. Therefore, the budget reflects 20% of the sites will require an initiation visit (for those unable to attend the Investigators’ Meeting).
Clinical Trial Agreements for Sites, Central Laboratory, and EDC Vendor
Medpace will prepare and provide sample clinical trial agreements (including budgets) for the study sites. VIVUS will review and approve the final draft versions of the clinical trial agreements. The agreements will be distributed and negotiated with each site by Medpace (with final approval by VIVUS). Medpace will make payments to the clinical sites according to the VIVUS-approved schedule. All payments for sites, Medpace Reference Laboratories, and Phase Forward will be made electronically to Medpace within seven days of invoice receipt. If electronic payment exceeds or falls below actual costs, VIVUS will adjust its payment based on the prior month’s payment reconciliation. Investigator payment invoices will include the following detail:
· Clinical study number;
· PI or Site #;
· Patient ID;
· Amounts paid per visit;
· Total amount earned to date;
· Prior payments; and
· Current payment amount.
Institutional Review Board and Initial Essential Documents Packages
Medpace will select a central Institutional Review Board (IRB) and coordinate the initial submissions to the IRB. VIVUS must approve the central IRB selected. Medpace will be responsible for payments to the central IRB selected. Medpace will be responsible for payments to the central IRB utilizing funds provided in the same manner as described above.
A study-specific, prototype informed consent form (ICF) will be designed by Medpace. The ICFs will be reviewed and approved by VIVUS. Medpace will distribute the ICFs to the Central IRB. Medpace will be responsible for negotiating changes to the informed consents with the central IRB.
Deviations from the VIVUS template must be brought to the attention of the VIVUS Clinical Leader who will facilitate VIVUS legal review and approval, if required.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
All components of the Initial Essential Documents Package will be collected, tracked, and maintained by Medpace according to Medpace SOPs. The Medpace Study Start-up team will review all documents, negotiate any changes with study site personnel, and correct any errors. The Initial Essential Document Package includes the following:
· Signed protocol signature pages;
· Financial disclosure questionnaires (FDQs) (template to be provided by VIVUS, Medpace to collect the forms);
· Clinical study agreement (includes study budget);
· FDA Form 1572;
· Laboratory certifications and reference values;
· Curricula vitae for all Investigators;
· IRB approval of the protocol (and any amendments), the informed consent and sponsor approved advertisements; and
· Qualification of IRB members.
The FDQs shall apply throughout the entire term of the study and for one year following last patient last visit (LPLV). If there is any change in the accuracy of a particular site’s FDQ during that time period, that site will be responsible for notifying Medpace of the change. If a site notifies Medpace of a change in staff from LPLV to one year after LPLV, Medpace will collect an updated FDQ and forward on to VIVUS. Costs associated with this task are included in the budget. Once a study has ended, Medpace sends a fax to all sites notifying them of study end as well as reminding them of their responsibilities (one of which being to notify Medpace of any FDQ changes).
Site Initiation Visits
Site initiation visits will be conducted by the Medpace CRAs consistent with Medpace SOPs. For purposes of this proposal, it is assumed that the Investigators’ Meeting will serve as the initiation visit and only 20% of the sites for each study will require separate initiation visits. Typically, if the Investigator Meeting is considered the initiation visit, the CRA will contact the site via phone to review study procedures and the CRA’s first routine monitoring visit will occur shortly (can be defined as 1 or 2 weeks) after a patient is screened for the trial. During this visit, the CRA will review bullets 2-5 below. A site initiation visit report will be completed and forwarded to VIVUS within 7 business days of the visit. These visits will include, but are not limited to, the following tasks:
· Train site and applicable study personnel on the protocol and study procedures;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Ensure the site has received all study supplies required for the conduct of the study, including study medication and access to eCRFs;
· Provide and review the Trial Master File binder. Medpace CRAs will provide instruction to the site personnel on the organization and maintenance of the documents in the binder;
· Review study medication accountability procedures;
· Provide eCRF completion instructions; and
· Explain the serious adverse event (SAE) reporting procedures.
CLINICAL OPERATIONS
Monitoring Data Review Guidelines
The Medpace Lead CRA in collaboration with the project team members will develop a project-specific Monitoring Plan for the study. This plan will include detailed interpretations of study expectations for the CRAs assigned to the study. Issues are discussed and updated on an ongoing basis throughout the project. Medpace will request that VIVUS approve the initial document and then re-approve the document on a quarterly basis.
Routine Clinical Monitoring Visits
Medpace will conduct routine monitoring visits at each site consistent with Medpace SOPs. The frequency of the visits will be determined by the site’s activity, but will be conducted on average every four to six weeks. Visits at the beginning and end of the study may be more frequent based on the needs of the study, including, but not limited to, recruitment, quality data and study close-out activities. Based on recruitment being very rapid, the first routine visit will be performed within 2 weeks after the first two patients are screened at the site. The CRA will perform 100% source documentation. In addition, data queries will be resolved during the visits, eCRF changes will be verified, and supporting documentation for SAEs will be obtained. The Medpace CRA will verify all laboratory samples have been obtained according to guidelines and the results are available in the patient’s source documents. A monitoring visit report will be forwarded to VIVUS within 10 business days of the visit. VIVUS will be notified of any significant issues by phone within one business day.
The following tasks will also be performed:
· Train any new site personnel and review study issues with applicable site personnel;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Ensure the site has sufficient study supplies (including study medication);
· Ensure the site is entering eligible patients into the study in a timely manner, and notify the Medpace Study Manager immediately of any problems;
· Detect any significant compliance or other issues and notify the Medpace Study Manager by phone, within one business day of the monitoring visit;
· Confirm the Trial Master File is complete and current, and the site is complying with applicable regulations and the protocol. VIVUS will be notified immediately of any significant deviations;
· Ensure the site is completing eCRFs in a timely manner;
· Ensure all completed eCRFs are reviewed, verified, corrected, and transmitted to Medpace;
· Review eCRFs for accuracy and protocol adherence;
· Verify study medication dispensing, compliance, and accountability for each patient; and
· Ensure the Investigator reported all SAEs to Medpace and the applicable IRB.
Medpace will provide a follow-up letter to the study site after each visit. The letter will include, but will not be limited to, the following:
· Important findings during the visit;
· Recommendations of corrective actions to be taken by the site; and
· Follow-up information regarding questions asked during the visit.
The Monitoring Visit Reports with all attachments including follow-up letters, will be available for view through the Medpace web based Clintrak® Study Management system within 10 business days of the monitoring visit.
In-house Clinical Monitoring Activities
Investigators will be contacted on a regular basis (every week during the active recruitment period and between monitoring visits) to ensure progress at the study site. The CRA will take the opportunity to review enrollment, answer protocol-related questions, discuss eCRF completion issues, obtain information regarding AEs, and ensure the site continues to be committed to the completion of the study in a timely manner and according to the protocol.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Telephone contacts will be entered in the Medpace ClinTrak Study Management system. Contacts requiring urgent attention will be relayed to VIVUS immediately and will be resolved in collaboration with the Medpace CTM and the VIVUS Study Manager.
Withdrawals Due to Adverse Events
Withdrawals due to AEs will be tracked and reconciled with the eCRF database on an ongoing basis by Medpace. The CRA will be responsible for reporting withdrawals due to AEs to VIVUS using the monitoring visit report.
Medpace will write narratives for all withdrawals due to AEs, for use in the clinical trial study report.
Status Reporting
The Medpace Senior CTM will serve as the central channel for communication between Medpace and VIVUS. The OB-301 CTM will work in conjunction with the clinical monitoring group to track study progress and report to VIVUS on a weekly basis. In addition, the OB-301 CTM will be responsible for overall management of site information, overseeing the status of Investigator contracts, direct supervision of CRAs, tracking of enrollment information, and distribution of study supplies. The Medpace OB-301 CTM will be the primary contact for the sites to address protocol interpretations and inclusion/exclusion criteria. All protocol-related issues will be recorded in an ongoing document to ensure consistency. The CTM is available 24 hours a day, 7 days a week via the Medpace Project Helpline.
Medpace will develop a communication plan at the start-up of the study. The CTM will work with the VIVUS project team to define details regarding study communication, including status reporting and team conference calls. Medpace typically delivers status reports on a predetermined day (and time) of the week, which is established around the weekly team calls so that the information can be discussed. Utilization of IVRS will also allow the project team to review patient status on a real time basis.
The Medpace CTM will collaborate with the Medpace Medical Expert and the VIVUS Study Manager to address any questions that may arise. The ClinTrak Study Management databases will serve as the primary source of project status information and will allow the Medpace CTM to report on any aspect of the study. The databases are updated on a real-time basis, providing accurate and up-to-date information.
Elements of ClinTrak include:
· Phone contacts;
· Monitoring visit reports;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Patient status including details on withdrawals during the treatment phase;
· Study supplies; and
· Protocol deviations.
Medpace will provide weekly status reports via a secure project website, to include the following status by site:
· Number of patients screened;
· Number of screen failures;
· Number of patients randomized;
· Number of patients dropped with drop rates; and
· Number of patients completed.
In addition, monthly reports will be provided, to include the following:
· Monitoring visits scheduled; and
· Monitoring visits completed.
Data Management status reports will be provided monthly via a secure website. These reports will include the following:
· Cumulative and interval eCRF status by site (including number of eCRFs transmitted and cleaned);
· Cumulative and interval patient status by site (including number of patients ongoing, completed, and early terminations) based on eCRF data in-house; and
· Cumulative and interval query status by site (including number of queries issued and days outstanding).
Project Website
Medpace will develop a secure OB-301 website that will be available to all project team members and site personnel. The website will include information and tools relevant to the study, such as status reports, meeting agendas and minutes, newsletters, monitor visit status, and the project timeline. Access is controlled by the type of user. Access to the tabs (sections) on the websites are controlled by the user type so that sites can have access to the section specifically designed for site access. Medpace can set up an automatic notification process of updates to the user email accounts. Clinical sites will have access only to parts of the website that pertain to their function. They will have the ability to receive study information and download study-related forms.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Team Meetings
VIVUS and Medpace
Medpace assumes that one face-to-face meeting, other than the Investigators’ meeting and kickoff meeting will take place for the OB-301 study at VIVUS.
Weekly teleconferences will be held during the study. The teleconferences will be held to discuss study progress and review project documents (as necessary). The Medpace Project Coordinators will be responsible for preparing and distributing agendas and minutes for each meeting/teleconference.
Additional meetings/teleconferences will be scheduled throughout the project, as needed.
Internal Meetings
Medpace will develop a project-specific internal project development/training program for all project team members. Included in this program will be the following:
· Protocol/eCRF review meeting;
· Medical in-services;
· Periodic Monitoring Plan meetings; and
· Periodic project meetings.
Newsletter
Medpace will prepare a 2-4 page full color site monthly newsletter for OB-301 as an additional avenue of communication and training for all site personnel. VIVUS will provide input and approval of the newsletter prior to distribution. Medpace will be responsible for printing and distributing two copies of each newsletter to each site.
Closeout Visits
Medpace will conduct closeout visits at each site consistent with Medpace SOPs after all patients have completed or discontinued from the study at the respective site. This visit may be performed as part of a final routine site monitoring visit. Site closeout visit reports will be forwarded to VIVUS within 10 business days of the visit. The following tasks will be performed:
· Resolve outstanding data queries;
· Ensure all study medication supplies are accounted for and that medication records and unused supplies are returned to VIVUS;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Ensure the Investigator’s copies of data and source documents are properly stored;
· Ensure the Trial Master File is complete, correct, and properly stored;
· Ensure the Investigator is aware of record retention requirements and other obligations, and a final site status report is sent to the IRB and VIVUS; and
· Instruct the site to update the FDQs for one year after the study is completed.
Site Audits
Site audit visits will be conducted, as deemed necessary, by VIVUS.
Medpace will respond to any audit findings and ensure the proper actions are taken to resolve outstanding issues.
REGULATORY AFFAIRS AND SAFETY REPORTING
Serious Adverse Events
All SAEs will be reported immediately, within 24 hours of discovery or notification of the event, by the clinical study site to VIVUS, or its designee, according to SOPs specified by VIVUS.
VIVUS will be responsible for submitting all immediately reportable SAEs (serious, causally related and unexpected) to the Food and Drug Administration (FDA) in accordance with the current regulations.
If a SAE has occurred at a site, the Medpace CRA will always 100% source document verify the event during the monitoring visit to ensure it has been recorded, documented, and reported appropriately and accurately. In addition, data queries will be resolved during the visits, CRF changes will be verified, and supporting documentation for SAEs will be obtained.
Medpace will provide VIVUS listings of non-serious adverse events from all sites to support filing of the Annual Safety Reports. Medpace will reconcile SAE listings with the AE database.
DATA MANAGEMENT
Data Management activities performed by Medpace will include eCRF tracking, preparation of a data management manual, eCRF review, coding of adverse events and concomitant medications, medical histories, data cleaning/editing, querying, query
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
tracking, final database quality review, and delivery of the final SAS® database. The data cleaning process will be performed on an ongoing basis following Medpace Data Management SOPs. Two data transfers (SAS transport files) will be performed: a test transfer prior to FPFV and a final transfer. A unit price for additional data transfers has been provided in the budget.
Database Development and Data Management Manual
Medpace will design and validate the data entry systems prior to entry of data. A Data Management Manual will be prepared for each study using the Medpace template, and will include the following data management documents:
· Database specifications, based on VIVUS specifications;
· Guidelines for the tracking of eCRFs and data queries;
· Data Management Guidelines, which will include guidelines for reviewing the data, and description of the database edit check specifications to be performed for data cleaning; and
· Description of the database quality control (QC) plan.
The manuals will be reviewed and approved by VIVUS.
Data from the Central Laboratory
Medpace will arrange periodic data transfers from MRL. Medpace will track and reconcile discrepancies between the MRL demographic data and the eCRF database, which are generated during the data cleanup process throughout each project.
Data Entry and Data Querying
Medpace assumes no codable forms will be transmitted for screen failure patients. Data is entered by site personnel that have been trained on the eCRF system. Data will be reviewed according to Medpace Data Management Guidelines and edits. A data query will be generated electronically within the eCRF system. The resolutions/corrections are made by site personnel by changing the data. All changes are recorded in an audit trail. All answered queries are verified/closed by Medpace Data Management. All resolutions/corrections will be performed consistent with Medpace SOPs. The Data Coordinators will work directly with the site personnel in resolving queries.
Coding
Medpace will be responsible for coding adverse events, medical histories, and concomitant medications.
· MedDRA will be used to code adverse events and medical histories. Adverse events and medical histories will be coded to the lowest level term, preferred term, and system organ class.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· WHO DRUG will be used to code concomitant medications. Concomitant medications will be coded to the generic name and anatomic therapeutic class 3. It is assumed by Medpace that VIVUS holds a valid agreement with the Uppsala Monitoring Centre (UMC) for the WHO DRUG dictionary.
All coding will be done on a single version of each coding system (versions to be agreed upon by VIVUS). Medpace will provide the coding dictionaries.
Data Flow Chart
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
STATISTICAL ANALYSIS
Medpace will develop the data analysis plan (DAP) per the VIVUS/Medpace format and template. VIVUS will review and approve the DAP prior to initiation of programming. Included in the DAP is a detailed statistical methodology and programming specification for all statistical analyses, tables/figures/listings (TFLs), and derived datasets.
Medpace will be responsible for programming and generating all TFLs, according to the Medpace standard analysis validation and quality control procedures. All TFLs will be programmed using SAS (Version 8), according to the VIVUS Programming Standards document. Pre-final TFLs will be generated twice on clean data for format review. Final TFLs will be generated on final data for the clinical study report.
MEDICAL WRITING
The Medpace Medical Writing team works in collaboration with the Medpace Medical Expert to provide a clinical study report according to FDA/ICH guidelines. The preparation of the Integrated Clinical/Statistical Study Report involves three stages of development: (1) the Study Report Shell (SRS), (2) the Pre-Final Study Report (PFSR), and (3) the Final Study Report (FSR).
The SRS is prepared after sign-off of the Final DAP. The SRS is created using a template and/or style guide provided by VIVUS, or by utilizing the Medpace standard report template, which adheres to the ICH guideline, “Structure and Content of Clinical Study Reports,” and follows the American Medical Association Manual of Style. The SRS incorporates information from the protocol, amendments, and eCRFs into sections of the report, including but not limited to the study design, study population, treatments administered, and the evaluation schedule. The statistical methods and results sections encompass information derived from the Final DAP. The results sections include mock-up in-text tables and text. The SRS undergoes a complete team review, which includes the Medical Monitor, Statistician, CTM, Data Manager, and other team members, if applicable. After the Medical Writer incorporates all team changes into the SRS, the SRS undergoes Medpace’s comprehensive document QC process. Once all changes from the document QC process are implemented into the SRS, the SRS is forwarded to the VIVUS for review.
Preparation of the PFSR occurs after receipt and implementation of VIVUS comments on the SRS, declaration of a clean database, and completion of Pre-Final Analyses. If necessary, a results review meeting is conducted with key members of the Medpace and VIVUS project team as the Medical Writer begins preparing the PFSR. The PFSR is a complete version of the report without the appendices. The PFSR undergoes a complete
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
team review, which includes the Medical Monitor, Statistician, CTM, Data Manager, and other team members, if applicable. After the Medical Writer incorporates all team changes into the PFSR, the PFSR undergoes Medpace’s comprehensive document QC process. Once all changes from the document QC process are implemented into the PFSR, the PFSR is forwarded to VIVUS for review.
The FSR is prepared once VIVUS’ comments on the PFSR are returned and all requested changes are agreed upon (including the acceptance of final text for all complicated or sensitive sections, which may require sending non-QC’d drafts of the report to VIVUS), and the Final Analyses are completed. The FSR is a complete version of the report including paginated appendices and/or supplements. The FSR undergoes a complete internal QC review and is forwarded to VIVUS for sign-off. The signed cover sheet is returned by VIVUS to verify their acceptance of the FSR.
STUDY CLOSEOUT
At the conclusion of each study, once all deliverables have been met, Medpace will return the original study files to VIVUS. These will include:
· General project administration files (this file includes items such as: Outside Vendor Correspondence, Multiple Site Correspondence (e.g. faxes to all sites), Central IRB Correspondence, Project Specific SOPs and Procedures, Monitoring Plan, Trial Master File, Project Timelines, Newsletters, and Meeting Minutes)
· Site files (this file includes site items such as: Essential Documents, Budget, Site Correspondence, Monitoring Visit Reports, Drug shipment documents, Study Supply forms, and Protocol Deviations);
· Final statistical tables and listings with results;
· Data management documentation (this file includes items such as: Database Definition Document, Final eCRFs, External Database Import Specs., Data Management Plan, Coding Reports, Analysis Plan, and Randomization Code); and
· Final clinical study report.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Assumptions
|
OB-301 |
|
Description |
|
Number of Investigators |
|
*** |
|
Number of Screened Patients |
|
*** |
|
Number of Randomized Patients |
|
*** |
|
Number of Randomized Patients/Site |
|
*** |
|
Duration of Enrollment Period |
|
*** |
|
Duration of Treatment Period |
|
*** |
|
Number of Investigators’ Meetings |
|
*** |
|
Number of Kickoff Meetings (at Medpace) |
|
*** |
|
Number of Sponsor Meetings (at VIVUS) |
|
*** |
|
Number of Conference Calls |
|
*** |
|
Frequency of Calls |
|
*** |
|
Number of Clinical Monitors |
|
*** |
|
Number of Pre-study Visits |
|
*** |
|
Number of Initiation Visits |
|
*** |
|
Number of Routine Monitoring Visits |
|
*** |
|
Number of Closeout Visits |
|
*** |
|
Monitoring Frequency |
|
*** |
|
Number of Newsletters per Site (monthly) |
|
*** |
|
Estimated Number of eCRFs per Completed Patient |
|
*** |
|
Estimated Number of Unique eCRFs per Completed Patient |
|
*** |
|
Total Number of eCRFs |
|
*** |
|
Estimated Number of AE Codes Per Patient |
|
*** |
|
Estimated Number of Medical History Codes Per Patient |
|
*** |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
Estimated Number of Concomitant Medications Codes Per Patient |
|
*** |
|
Estimated Number of Queries Per Patient |
|
*** |
|
External Data Sources |
|
*** |
|
Data Transfers from Medpace to VIVUS |
|
*** |
|
Number of Raw Listings |
|
*** |
|
Number of Unique TFs |
|
*** |
|
Number of Version TFs |
|
*** |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #01
Appendix 5 — Project Schedule
VIVUS, Inc.
Qnexa ▇▇-▇▇▇
▇▇▇▇▇▇▇▇▇▇
|
▇▇-▇▇▇ |
|
Date |
|
Medpace Begins Work |
|
*** |
|
Protocol Finalized |
|
*** |
|
First Patient First Visit |
|
*** |
|
Last Patient First Visit |
|
*** |
|
Last Patient Last Visit |
|
*** |
|
Final Database Lock |
|
*** |
|
Final TFLs Available |
|
*** |
|
Delivery of Final Clinical Study Report |
|
*** |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
CONFIDENTIAL |
September 11, 2007 |
Task Order #01
Appendix 4 — Budget
VIVUS, Inc.
Qnexa OB-301
Medpace Fee Estimate
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
CONFIDENTIAL |
September 11, 2007 |
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #01
Appendix 3 — Payment Schedule
VIVUS, Inc.
Qnexa OB-301
Payment Schedule
As set forth in this Agreement, professional service fees totalling $*** will be paid by VIVUS for professional services rendered by Medpace according to the following schedule.
***
Pass-through expenses will be billed to VIVUS on a monthly basis as incurred. The first Wire-Transfer payment will be invoiced prior to any site becoming active to ensure funds are available for site payments. Medpace will pay sites immediately following receipt of Wire-Transfers. Medpace will invoice VIVUS on a monthly basis and payments will be made by VIVUS within seven days of invoice receipt.
Payment Information and General Conditions
***
Inflation
The fees stipulated in the fee estimate include inflation for the duration of the study as specified in this proposal. Any significant shift in timelines will require a revision to the fees.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
CONFIDENTIAL |
September 7, 2007 |
|
Appendix 6 |
Transfer of Obligations Form |
CONFIDENTIAL
Directions: Complete a form for each clinical study where Sponsor obligations have been transferred in accordance with 21 CFR Part 312, Subpart D (Responsibilities of Sponsors). Forward the completed form to Sponsor’s Regulatory Affairs Department for submission to the applicable regulatory agencies.
|
Drug: |
VI-0521 |
|
Study ID: |
OB 301 |
|
Study Title: |
A phase III, randomized, double-blind, parallel design study comparing multiple doses of VI-0521 to placebo and their single-agent phentermine and topiramate constituents for the treatment of obesity in adults. | |||
|
CRO Name: |
Medpace, Inc. | |||
|
CRO Address: |
*** | |||
OBLIGATIONS TRANSFERRED TO MEDPACE: R THE APPROPRIATE BOX(ES).
o All obligations in 21 CFR 312, Subpart D (Responsibilities of Sponsors) have been transferred to Medpace.
x The following obligations have been transferred to Medpace:
Sec. 312.32: IND Safety Reports
x Promptly review safety information. *Sponsor will be notified within one (1) business day of discovery of significant new or serious adverse events or risks, or any unusual frequency of reactions with respect to the drug.
o Notify all participating investigators in a written IND safety report of any AE associated with the drug that is both serious and unexpected.
o Notify the FDA in a written IND safety report of any AE associated with the drug that is both serious and unexpected.
Sec. 312.53: Selecting investigators and monitors
x (a) Select qualified investigators
x (b) Control investigational drug shipment
x (c) Obtain information from investigators
x (1) Signed Form FDA-1572
x (2) CV or other qualification statement
x (3) Clinical protocol outline
x (4) Financial disclosure information
x (d) Select qualified monitors
Sec. 312.54: Emergency research
o (a) Monitor the progress of all studies involving an exception from informed consent.
o (b) Monitor such studies to identify when an IRB determines that it can’t approve the research.
Sec. 312.55: Informing investigators
x (a) Provide sites with the current Inv. Brochure.
x (b) Inform investigators of new observations on the drug, particularly with respect to AEs and safe use.
Sec. 312.56: Review of ongoing investigations
o (a) Monitor the progress of all IND studies.
x (b) Secure compliance from noncompliant investigators or discontinue drug shipments and end the investigator’s participation in the study.
o (c) Review and evaluate the safety and efficacy results as it is obtained from the investigator.
o (d) Discontinue use of the investigational drug if it is determined to present an unreasonable and significant risk to subjects, notify all IRBs and investigators, and assure the return or alternate disposition of the drug from the investigators.
Sec. 312.57: Record keeping and record retention
x (a) Maintain adequate records showing investigational drug receipt, shipment, or other disposition. *Master Drug Logs will include the name of the Investigator to whom the drug is shipped, the date, and the quantity and batch of each such shipment.
x (b) Maintain complete and accurate records showing any financial interests of the investigator subject to 21 CFR 54.
x (c) Retain the records and reports required by the regulations for 2 years after the marketing application is approved, or if not approved, until 2 years after investigational drug shipment is discontinued and FDA has been notified.
o (d) Retain reserve samples of any test article and reference standard identified and used in bioequivalence or bioavailability studies.
Sec. 312.58: Inspection of sponsor’s records and reports
x (a) Permit FDA personnel to have access to and copy and verify any records and reports related to the clinical investigation.
x (b) Permit DEA personnel to have access to and copy records related to the shipment, delivery, receipt and disposition of any investigational controlled substance. Assure adequate storage precautions are taken for investigational new drug substances listed in any schedule of the Controlled Substances Act.
Sec. 312.59: Disposition of unused supply of investigational drug
x Assure the return (or alternate disposition) of all unused supplies of the investigational drug from each discontinued/terminated investigator; maintain written records of any disposition of the investigational drug.
Other
o Please describe any other applicable transfers below:
September 7, 2007
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
MEDPACE
VIVUS Task Order #02
EXHIBIT A
Qnexa OB-302 TASK ORDER
MEDPACE Task Order Number: 02
MEDPACE Project Number: VOB302
This Task Order, dated September 12, 2007, is between Medpace, Inc. (“MEDPACE”), and VIVUS, Inc. (“VIVUS”).
RECITALS:
WHEREAS, MEDPACE and VIVUS have entered into that certain Master Services Agreement dated September 12, 2007 (the “Master Services Agreement”); and
WHEREAS, pursuant to the Master Services Agreement, MEDPACE has agreed to perform certain Services in accordance with Task Orders from time to time entered into by the Parties and VIVUS and MEDPACE now desire to enter into such a Task Order; and
WHEREAS, MEDPACE and VIVUS desire that MEDPACE provide certain services with respect to a phase III randomized, double blind, placebo controlled multicenter study to determine the safety and efficacy of VI-0521 in the treatment of obesity in otherwise healthy adults (the “Study”) for the study of the product VI-0521 (“Study Product”) as set out in the Protocol Number: OB-302, which is attached hereto as Appendix 1;
NOW, THEREFORE, in consideration of the mutual covenants contained herein, the Parties hereby agree as follows:
1. Scope of Work: MEDPACE shall perform the services described in the Scope of Work, attached hereto as Appendix 2, in accordance with the Project Schedule, attached hereto as Appendix 3 and any other documents attached to and specifically referenced in this Task Order (“Services”)
2. Compensation: For performance of these Services, VIVUS shall pay to MEDPACE an amount equal to the Project Budget set forth in Appendix 4, which amount shall be payable pursuant to the Payment Schedule set forth in Appendix 5.
3. Transfer of Obligations: Sponsor Obligations transferred to MEDPACE by VIVUS (consistent with the regulations set forth in 21 C.F.R. Section 312, Subpart D) are identified in Appendix 6.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
4. MSA. The provisions of the Master Services Agreement are hereby expressly incorporated by reference into and made a part of this Task Order.
IN WITNESS WHEREOF, the Parties have hereunto signed this Task Order effective as of the day and year first written above.
|
MEDPACE, INC. |
| ||
|
|
| ||
|
Signature: |
/s/ August ▇. ▇▇▇▇▇▇▇▇ |
| |
|
|
|
| |
|
By: |
August ▇. ▇▇▇▇▇▇▇▇ |
| |
|
|
(Print Name) |
| |
|
Title: |
President |
| |
|
|
|
| |
|
Date: |
September 12, 2007 |
| |
|
|
|
| |
|
|
|
| |
|
SPONSOR |
|
| |
|
|
|
| |
|
Signature: |
/s/ ▇▇▇▇▇▇ ▇. Day |
| |
|
|
|
| |
|
By: |
▇▇▇▇▇▇ ▇. Day |
| |
|
|
(Print Name) |
| |
|
Title: |
Vice President, Clinical Development |
| |
|
|
|
| |
|
Date: |
September 10, 2007 |
| |
List of Appendices:
Appendix 1: Protocol
Appendix 2: Scope of Work
Appendix 3: Project Schedule
Appendix 4: Project Budget
Appendix 5: Payment Schedule
Appendix 6: Transfer of Obligations
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
VI-0521
Protocol No. OB-302
***
CLINICAL PROTOCOL
A PHASE III RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED MULTICENTER STUDY TO DETERMINE THE SAFETY AND EFFICACY OF VI-0521 IN THE TREATMENT OF OBESITY IN AN ADULT POPULATION WITH BMI > ***
|
Compound: |
|
VI-0521 |
|
|
|
|
|
Compound Name (if applicable): |
|
Phentermine plus Topiramate |
|
|
|
|
|
US IND Number (if applicable): |
|
*** |
|
|
|
|
|
Protocol Number: |
|
OB 302 |
|
|
|
|
|
Phase: |
|
3 |
|
|
|
|
|
Medical Monitor: |
|
*** |
|
|
|
|
|
Sponsor: |
|
VIVUS, Inc. |
|
|
|
|
|
Version and Date: |
|
*** |
This document contains confidential information belonging to VIVUS, Inc. Except as otherwise agreed to in writing, by accepting or reviewing this document, you agree to hold this information in confidence and not copy or disclose it to others (except where required by applicable law) or use it for unauthorized purposes. In the event of any actual or suspected breach of this obligation, VIVUS, Inc must be promptly notified.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
VIVUS — Company Confidential
INTERNAL PROTOCOL APPROVAL
Protocol Number: OB-302
Title: A Phase III Randomized, Double-Blind, Placebo-Controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in an Adult Population with BMI > ***
The signature below documents that the reviewer has read and approved the attached protocol.
|
|
|
Signature |
|
Date |
|
|
|
|
|
|
|
Author ▇▇▇▇▇▇▇▇ ▇▇▇▇▇▇, PhD, MPH |
|
|
|
|
|
Clinical Consultant |
|
|
|
|
|
|
|
|
|
|
|
▇▇▇▇▇▇ ▇. Day, PhD |
|
|
|
|
|
VP, Clinical Development |
|
|
|
|
|
|
|
|
|
|
|
▇▇▇▇▇▇▇▇▇▇ ▇▇▇▇▇▇▇▇▇, PhD |
|
|
|
|
|
Sr. Director, Regulatory Affairs |
|
|
|
|
|
|
|
|
|
|
|
▇▇▇ ▇▇▇▇▇▇ |
|
|
|
|
|
▇▇. Director, Pharmaceutical Development |
|
|
|
|
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
PRINCIPAL INVESTIGATOR SIGNATURE
Protocol Number: OB-302
Title: A Phase III Randomized, Double Blind, Placebo-Controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in an Adult Population with BMI > ***
The signature below indicates that the principal investigator has read and understands the protocol and agrees to conduct the study in accordance with the protocol, applicable guidelines for Good Clinical Practices, the Declaration of Helsinki and all applicable regulatory guidelines and requirements. Please return one copy of this executed page to VIVUS, Inc.
|
Printed Name: |
|
|
|
|
|
|
|
Signature: |
|
Date: |
|
|
|
|
|
Facility Name: |
|
|
|
|
|
|
|
Address: |
|
|
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
PROTOCOL SYNOPSIS
Rationale:
Obesity leads to the development of co-morbidities such as hypertension, type 2 diabetes, dyslipidemia, coronary artery disease (CAD) and stroke. Weight reduction in obese individuals has been shown to delay or prevent the onset of these co-morbidities, or even reverse the damage caused by these co-morbidities. Diet, exercise and behavior modification therapy can be effective short-term treatments; however, many people experience difficulty in achieving and maintaining weight reduction without pharmacotherapy. VI-0521 is an investigational weight loss therapy that is a new combination of two currently approved drugs, phentermine and topiramate. This novel combination treatment may provide a safe and effective option for the achievement and maintenance of weight loss in obese adults.
Objectives:
The objectives of this study are to evaluate the safety and efficacy of two doses of VI-0521 for the treatment of obesity in obese adults with body mass index (BMI) > ***.
Trial Design:
The study is a randomized, double-blind placebo-controlled trial with subjects randomized to receive daily treatment with VI-0521 *** or *** or ***, with the total duration of treatment being 56 weeks. Randomization will be stratified by *** of subjects will be ***. Approximately 1250 subjects will be treated under the protocol with *** subjects randomized to *** and ***. Up to *** study sites in the USA will be employed.
Subjects will be instructed to follow a mild hypocaloric diet representing a 500-calorie/day deficit and to implement a lifestyle modification program, as tolerated, throughout the study period. During the first 4 weeks of treatment (weeks 1-4), study medication will be titrated to the assigned dose level, with the dosage increased each week as determined by randomization group. During treatment weeks 5-56, the dose will be maintained at the final dose level. Subjects who are unable to tolerate the assigned dosage may be treated at a reduced dose level or may take a drug holiday as defined in the protocol.
All subjects will return at approximately 4-week intervals for measurement and evaluation. Female subjects of child bearing potential will undergo a urine pregnancy test at each visit. Subjects who discontinue the treatment during the study will be encouraged to remain on study for all study-related procedures. Additionally, those who choose to discontinue the study will be encouraged to return at the 56-week time point for measurements and evaluation.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Study Subjects
The study population will consist of adult males and females up to 70 years of age with BMI ³***, fasting blood glucose ***. Female subjects of childbearing potential must agree to use adequate contraception (a double barrier method, stable hormonal contraception plus single barrier method or tubal ligation) for the duration of treatment.
Major exclusion criteria include: type 2 diabetes; known or suspected clinically significant valvular heart disease; clinically significant ECG abnormality; clinically significant hepatic or renal disease; clinically significant thyroid dysfunction, as evidenced by signs, symptoms, or TSH > 1.5 x ULN; obesity of known genetic or endocrine origin; history of bipolar disorder or psychosis, depression of moderate or greater severity, or presence or history of suicidal behavior or active suicidal ideation; recent weight instability; history of glaucoma or increased intraocular pressure; prior bariatric surgery; or smoking cessation within 3 months prior to enrollment.
Efficacy Endpoints:
The primary endpoints are the differences between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), and percent of subjects with weight loss of 5% or more at end of treatment (week 56). Percent weight loss will be calculated as ***.
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) between *** and *** groups;
· The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) between *** and *** groups; and
· The difference in change in waist circumference (randomization to week 56) between *** and *** groups.
Additional efficacy endpoints include:
· Effect on ***;
· Effect on body composition, as evaluated by ***. *** assessments will be performed at only at a ***.
· Effect on *** for *** and ***;
· The change in obesity-associated risk factors (total cholesterol, triglycerides, LDL-C, HDL-C, fasting glucose, blood pressure);
· Change from baseline to week 28 and week 56 in BMI;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Difference between *** and *** groups in the rate of progression to type 2 diabetes; and
· Baseline adjusted change in Framingham 10-year risk score at weeks 28 and 56.
The change in weight loss (percent, absolute, percent of subjects achieving weight loss of > 5% and > 10% of starting weight), waist circumference and obesity associated risk factors will also be assessed over time. Subgroup analyses, including but not limited to analysis by gender, age and race, may be performed.
***:
*** will also be evaluated. Data will be obtained using a multiple trough sampling scheme with samples collected at *** and ***. Effects of various cofactors including (but not limited to) ***, gender, race, ***, and age will be evaluated.
Safety Endpoints:
Safety will be assessed by an evaluation of adverse events, including eye symptoms (collected each study visit); ***. All subjects will be screened for the *** using a validated survey instrument ***, and for *** using the ***. Follow-up assessments will be done at *** after ***.
Statistical Methods:
All subjects who are randomized, take one or more doses of test material and have at least one post treatment measurement will be included in the analysis. Comparisons between treatments will be assessed using a *** with factors of *** and *** and with *** for percent weight loss, and by *** for percent of subjects achieving at least 5% weight loss. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant at *** for both co-primary endpoints, then the test will proceed to the *** also at the ***. If the statistical comparison is not significant at the ***, then the statistical test will be stopped and the *** will not be tested. If both dose groups are significantly better than ***, then the two active dose groups will be compared. A *** of difference in response rate between treatment groups will be derived. The *** for subjects who discontinue treatment prior to completion of the study, ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
TABLE OF CONTENTS
|
INTERNAL PROTOCOL APPROVAL |
|
2 | |
|
|
|
| |
|
PRINCIPAL INVESTIGATOR SIGNATURE |
|
3 | |
|
|
|
| |
|
PROTOCOL SYNOPSIS |
|
4 | |
|
|
|
| |
|
TABLE OF CONTENTS |
|
7 | |
|
|
|
|
|
|
1 |
INTRODUCTION |
|
13 |
|
|
|
|
|
|
1.1 |
Background |
|
13 |
|
|
|
|
|
|
1.2 |
Rationale |
|
14 |
|
|
|
|
|
|
2 |
TRIAL OBJECTIVES |
|
14 |
|
|
|
|
|
|
3 |
TRIAL DESIGN |
|
14 |
|
|
|
|
|
|
4 |
SUBJECT SELECTION |
|
16 |
|
|
|
|
|
|
4.1 |
Inclusion Criteria |
|
16 |
|
|
|
|
|
|
4.2 |
Exclusion Criteria |
|
16 |
|
|
|
|
|
|
4.3 |
Randomization Criteria |
|
18 |
|
|
|
|
|
|
4.4 |
Life Style Guidelines |
|
18 |
|
|
|
|
|
|
5 |
TRIAL TREATMENTS |
|
19 |
|
|
|
|
|
|
5.1 |
Allocation to Treatment |
|
19 |
|
|
|
|
|
|
5.2 |
Breaking the Blind |
|
19 |
|
|
|
|
|
|
5.3 |
Drug Supplies |
|
20 |
|
|
|
|
|
|
5.3.1 |
Formulation and Packaging |
|
20 |
|
|
|
|
|
|
5.3.2 |
Preparation and Dispensing |
|
20 |
|
|
|
|
|
|
5.3.3 |
Administration |
|
20 |
|
|
|
|
|
|
5.3.4 |
*** |
|
21 |
|
|
|
|
|
|
5.3.5 |
Compliance |
|
21 |
|
|
|
|
|
|
5.4 |
Drug Storage and Drug Accountability |
|
21 |
|
|
|
|
|
|
5.5 |
Concomitant Medication(s) |
|
22 |
|
|
|
|
|
|
5.5.1 |
Excluded Medications |
|
22 |
|
|
|
|
|
|
5.5.2 |
Other Restricted Medications |
|
22 |
|
|
|
|
|
|
5.5.3 |
Documentation of Concomitant Medication Use |
|
22 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
5.6 |
Treatment of Diabetes |
|
22 |
|
|
|
|
|
|
5.7 |
Treatment of *** |
|
23 |
|
|
|
|
|
|
5.8 |
Treatment of *** |
|
23 |
|
|
|
|
|
|
6 |
TRIAL PROCEDURES |
|
23 |
|
|
|
|
|
|
6.1 |
Screening (Visit 1) |
|
24 |
|
|
|
|
|
|
6.2 |
Treatment Period |
|
24 |
|
|
|
|
|
|
6.2.1 |
Randomization (Visit 2) |
|
24 |
|
|
|
|
|
|
6.2.2 |
Titration (Visit 3) |
|
25 |
|
|
|
|
|
|
6.2.3 |
Treatment (Visits 4 through 16) |
|
26 |
|
|
|
|
|
|
6.2.4 |
End of Treatment (Visit 17 or withdrawal from study) |
|
27 |
|
|
|
|
|
|
6.3 |
Study Period |
|
27 |
|
|
|
|
|
|
6.4 |
Subject Withdrawal |
|
28 |
|
|
|
|
|
|
7 |
ASSESSMENTS |
|
29 |
|
|
|
|
|
|
7.1 |
Weight, Waist Circumference, Height and BMI Assessment |
|
29 |
|
|
|
|
|
|
7.1.1 |
Weight Assessment |
|
29 |
|
|
|
|
|
|
7.1.2 |
Waist Circumference Measurement |
|
29 |
|
|
|
|
|
|
7.1.3 |
Height and BMI |
|
30 |
|
|
|
|
|
|
7.2 |
▇▇▇▇▇ ▇▇▇▇▇ |
|
30 |
|
|
|
|
|
|
7.3 |
Questionnaires |
|
31 |
|
|
|
|
|
|
7.3.1 |
*** |
|
31 |
|
|
|
|
|
|
7.3.2 |
*** |
|
31 |
|
|
|
|
|
|
7.3.3 |
*** |
|
31 |
|
|
|
|
|
|
7.4 |
*** and End of Treatment Questions |
|
31 |
|
|
|
|
|
|
7.4.1 |
*** |
|
31 |
|
|
|
|
|
|
7.4.2 |
End of Treatment Questions |
|
31 |
|
|
|
|
|
|
7.5 |
Laboratory Tests |
|
32 |
|
|
|
|
|
|
7.5.1 |
Blood Chemistry |
|
32 |
|
|
|
|
|
|
7.5.2 |
Hematology |
|
32 |
|
|
|
|
|
|
7.5.3 |
Urinalysis |
|
32 |
|
|
|
|
|
|
7.5.4 |
Biomarkers |
|
32 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
7.5.5 |
Urine Pregnancy Test |
|
33 |
|
|
|
|
|
|
7.5.6 |
Thyroid Stimulating Hormone (TSH) |
|
33 |
|
|
|
|
|
|
7.5.7 |
Antibody to HIV, HCV, HBsAg |
|
33 |
|
|
|
|
|
|
7.5.8 |
Urine Drug Screen |
|
33 |
|
|
|
|
|
|
7.5.9 |
Screening Serum Sample (Retain) |
|
33 |
|
|
|
|
|
|
7.6 |
*** Evaluation |
|
33 |
|
|
|
|
|
|
7.7 |
Physical Examination |
|
33 |
|
|
|
|
|
|
7.8 |
Electrocardiogram (ECG) |
|
34 |
|
|
|
|
|
|
7.9 |
Framingham Risk Score |
|
34 |
|
|
|
|
|
|
7.10 |
Body Composition |
|
34 |
|
|
|
|
|
|
8 |
ADVERSE EVENTS |
|
35 |
|
|
|
|
|
|
8.1 |
Adverse Events |
|
35 |
|
|
|
|
|
|
8.1.1 |
Severity Assessment |
|
35 |
|
|
|
|
|
|
8.1.2 |
Causality Assessment |
|
35 |
|
|
|
|
|
|
8.1.3 |
Abnormal Test Findings |
|
36 |
|
|
|
|
|
|
8.2 |
Serious Adverse Events |
|
36 |
|
|
|
|
|
|
8.2.1 |
Definition of Hospitalization |
|
37 |
|
|
|
|
|
|
8.3 |
Eliciting Adverse Event Information |
|
37 |
|
|
|
|
|
|
8.3.1 |
Eye Pain |
|
37 |
|
|
|
|
|
|
8.3.2 |
*** |
|
38 |
|
|
|
|
|
|
8.4 |
Reporting Period |
|
38 |
|
|
|
|
|
|
8.5 |
Reporting Requirements |
|
38 |
|
|
|
|
|
|
8.5.1 |
Serious Adverse Event Reporting Requirements |
|
38 |
|
|
|
|
|
|
8.5.2 |
Non-Serious Adverse Event Reporting Requirements |
|
39 |
|
|
|
|
|
|
8.5.3 |
*** |
|
39 |
|
|
|
|
|
|
9 |
DATA ANALYSIS/STATISTICAL METHODS |
|
39 |
|
|
|
|
|
|
9.1 |
Sample Size Determination |
|
39 |
|
|
|
|
|
|
9.2 |
Efficacy Analysis |
|
39 |
|
|
|
|
|
|
9.2.1 |
Analysis of Primary Endpoint |
|
39 |
|
|
|
|
|
|
9.2.2 |
Analysis of Secondary Endpoints |
|
40 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
9.3 |
Analysis of Other Endpoints |
|
41 |
|
|
|
|
|
|
9.3.1 |
Other Efficacy Endpoints |
|
41 |
|
|
|
|
|
|
9.3.2 |
*** Analysis |
|
41 |
|
|
|
|
|
|
9.4 |
Analysis Populations |
|
41 |
|
|
|
|
|
|
9.5 |
*** |
|
42 |
|
|
|
|
|
|
9.6 |
*** |
|
42 |
|
|
|
|
|
|
9.7 |
Safety Analysis |
|
42 |
|
|
|
|
|
|
9.7.1 |
Adverse Events |
|
42 |
|
|
|
|
|
|
9.7.2 |
Clinical Laboratory Tests |
|
42 |
|
|
|
|
|
|
9.7.3 |
▇▇▇▇▇ ▇▇▇▇▇ and Other Safety Evaluations |
|
42 |
|
|
|
|
|
|
9.7.4 |
Questionnaire Assessments |
|
43 |
|
|
|
|
|
|
9.8 |
Interim Analysis |
|
43 |
|
|
|
|
|
|
9.9 |
*** |
|
43 |
|
|
|
|
|
|
10 |
QUALITY CONTROL AND QUALITY ASSURANCE |
|
43 |
|
|
|
|
|
|
11 |
DATA HANDLING AND RECORD KEEPING |
|
43 |
|
|
|
|
|
|
11.1 |
Case Report Forms / Electronic Data Record |
|
43 |
|
|
|
|
|
|
11.2 |
Record Retention |
|
44 |
|
|
|
|
|
|
12 |
ETHICS |
|
44 |
|
|
|
|
|
|
12.1 |
Institutional Review Board (IRB)/Independent Ethics Committee (IEC) |
|
44 |
|
|
|
|
|
|
12.2 |
Ethical Conduct of the Trial |
|
45 |
|
|
|
|
|
|
12.3 |
Subject Information and Consent |
|
45 |
|
|
|
|
|
|
12.4 |
Disclosure of Data |
|
45 |
|
|
|
|
|
|
13 |
REGULATORY CORRESPONDENCE |
|
46 |
|
|
|
|
|
|
14 |
DEFINITION OF END OF TRIAL |
|
46 |
|
|
|
|
|
|
15 |
SPONSOR DISCONTINUATION CRITERIA |
|
46 |
|
|
|
|
|
|
16 |
PUBLICATION OF TRIAL RESULTS |
|
47 |
|
|
|
|
|
|
17 |
REFERENCES |
|
48 |
|
|
|
|
|
|
APPENDIX 1: SCHEDULE OF STUDY ACTIVITIES |
|
51 | |
|
|
|
|
|
|
APPENDIX 2: *** |
|
52 | |
|
|
|
| |
|
APPENDIX 3: *** |
|
55 | |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
APPENDIX 4: PROTOCOL AMENDMENTS |
|
57 |
|
|
|
|
|
TABLES |
|
|
|
|
|
|
|
Table 1: VI-0521 Dosage Strengths by Titration Week for Each Treatment Group |
|
20 |
|
|
|
|
|
FIGURES |
|
|
|
|
|
|
|
Figure 1. Schematic Representation of Study Design |
|
15 |
|
|
|
|
|
Figure 2. Measuring Tape Position for Waist Circumference Assessments |
|
30 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
LIST OF ABBREVIATIONS
|
ACE |
|
angiotensin converting enzyme |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
Cl |
|
apparent clearance |
|
cm |
|
centimeters |
|
CRFs |
|
case report forms |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
DSM-IV |
|
Diagnostic and Statistical Manual IV |
|
ECG |
|
electrocardiogram |
|
FDA |
|
Food and Drug Administration |
|
*** |
|
*** |
|
GCP |
|
Good Clinical Practices |
|
*** |
|
*** |
|
HBsAg |
|
hepatitis B surface antigen |
|
HCV |
|
hepatitis C virus |
|
HDL-C |
|
high density lipoprotein cholesterol |
|
HIV |
|
human immunodeficiency virus |
|
ICH |
|
International Conference on Harmonization |
|
IEC |
|
Independent Ethics Committee |
|
IND |
|
Investigational New Drug |
|
IRB |
|
Institutional Review Board |
|
ITT |
|
intent to treat |
|
IVRS |
|
Interactive Voice Response System |
|
*** |
|
*** |
|
kcal/day |
|
kilocalories per day |
|
kg |
|
kilogram |
|
kg/m2 |
|
kilogram per square meter |
|
*** |
|
*** |
|
LDL-C |
|
low density lipoprotein cholesterol |
|
µU/mL |
|
microunits per milliliter |
|
mg |
|
milligram |
|
mg/day |
|
milligram per day |
|
mg/dL |
|
milligram per deciliter |
|
mmHg |
|
millimeters of mercury |
|
MedDRA |
|
Medical Dictionary for Regulatory Activities |
|
NYHA |
|
New York Heart Association |
|
*** |
|
*** |
|
*** |
|
*** |
|
QOD |
|
every other day |
|
RBC |
|
red blood cell |
|
SAE |
|
serious adverse event |
|
*** |
|
*** |
|
*** |
|
*** |
|
TSH |
|
thyroid stimulating hormone |
|
ULN |
|
upper limit of normal |
|
*** |
|
*** |
|
WBC |
|
white blood cells |
|
WHO |
|
World Health Organization |
|
WNL |
|
within normal limits |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
1 INTRODUCTION
VI-0521 is an investigational weight loss therapy that is a combination of two approved drugs, phentermine and topiramate. Phentermine hydrochloride (phentermine), an anorectic agent, has sympathomimetic actions and stimulates the central nervous system.(1),(2) It is postulated that ***, and there is a ***.(3), (4) Topiramate, an anticonvulsant agent that has been shown to induce weight loss, is known to ***.(5) However, the specific mechanism mediating the weight loss is not known. The present study is being conducted to evaluate the combined use of phentermine and topiramate at doses of 3.75 mg phentermine/23 mg topiramate and 15 mg phentermine/92 mg topiramate in the treatment of obesity in adult subjects with body mass index (BMI) greater than or equal to ***.
1.1 Background
Recent studies have shown that about 129 million adults in the United States are clinically overweight or obese.(6) In recent years, there has been a dramatic increase in obesity in both children and adults.(7), (8) Results from the National Health and Nutrition Examination Survey showed an overall 32.2% prevalence of obesity during 2003-2004, with obesity increasing from 27.5% in 1999-2000 to 31.1% in 2003-2004 in men but remaining relatively unchanged in women (1999-2000, 33.4%; 2003-2004, 33.2%).(8)
Obesity is associated with numerous co-morbidities including dyslipidemia, coronary artery disease, hypertension, stroke and type 2 diabetes.(7), (9) Epidemiological data indicate that obesity is associated with increased mortality,(10) and a recent study of over 500,000 individuals concluded that excess body weight during midlife was associated with an increased risk of death.(11) A modest weight loss (5-10%) can result in a marked reduction in obesity-related metabolic and cardiovascular risk factors.(12), (13), (14) Diet, exercise and behavior modification are standard treatments for obesity although most obese individuals do not achieve prolonged weight reduction without supplemental pharmacotherapy. However, the medications currently approved by the Food and Drug Administration (FDA) for weight loss are often poorly tolerated due to side effects and often fail to maintain long-term efficacy.(15)
Phentermine hydrochloride, a synthetic sympathomimetic amine, is an anorectic agent approved by the FDA as a short-term adjunct to a weight loss regimen based on exercise, behavior modification and caloric restriction. The usual adult dosage is *** administered either once daily or in divided doses.(1) The mechanism of action of phentermine for weight loss is similar to that of other anorectic agents; it decreases appetite and stimulates the central nervous system.(1) It is postulated that ***, and there is a ***.(3), (4) Additionally, increased *** levels may result in a decrease in *** that may result in increased satiety and decreased appetite.
Topiramate, a sulfamate-substituted monosaccharide, is an anticonvulsant agent indicated as adjunctive therapy for partial onset seizures, primary generalized tonic-clonic seizures, seizures
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
associated with Lennox-Gastaut syndrome and for migraine headache prophylaxis.(5) The recommended total daily dose of topiramate for treatment of seizures in adults is *** administered in two divided doses; ***. Topiramate is known to ***.(5) Recent clinical studies have shown that topiramate can promote weight loss in ***.(16), (17), (18) However, the exact mechanism by which topiramate exerts its anorectic effect is unknown although it is postulated to ***. A detailed summary of the clinical and nonclinical toxicology, pharmacokinetics and metabolism of phentermine and topiramate is provided in the Investigator Brochure.
VI-0521 has been studied in a Phase 2, randomized, double-blind clinical trial of 200 otherwise healthy obese adults under an investigator-initiated investigational new drug application (IND).(19) In this study, subjects were randomized into 1 of 4 treatment groups: ***. Treatment was continued for *** (including ***). Weight loss (percent of total body weight) among subjects treated with VI-0521 ***, compared to *** among *** among ***, and *** among ***. Subjects treated with VI-0521 *** than subjects in the other treatment groups. Significant decreases vs. *** and *** were observed in subjects receiving VI-0521. Of the 200 subjects randomized, *** completed treatment to ***; a *** completion rate was reported for subjects in the *** group. No deaths or serious adverse events were reported during the study and no significant changes in heart valve morphology were observed. The most commonly reported adverse events in subjects treated with VI-0521 were ***.
1.2 Rationale
VI-0521 is an investigational weight loss therapy that is a combination of two currently approved drugs, phentermine and topiramate. As such, VI-0521 represents a potential advance in the medical treatment of obesity since the two agents comprising this combination product effect weight loss through different mechanisms. Additionally, some of the expected side effects of the two drugs may be mitigated by complementary effects of the other. Thus, combining phentermine with topiramate may produce a similar or better adverse event profile compared to either of these agents individually. Topiramate therapy has been associated with ***. Due to the ***, it is mechanistically possible that ***. Thus, the dual mechanisms and low drug doses employed in VI-0521 may provide a safe and effective pharmacotherapy for the achievement and maintenance of weight loss in obese adults.
2 TRIAL OBJECTIVES
The objectives of this study are to evaluate the safety and efficacy of two doses of VI-0521 for the treatment of obesity in adults with body mass index (BMI) > ***.
3 TRIAL DESIGN
In this prospective, randomized, double blind, placebo-controlled trial, subjects meeting the eligibility criteria will be randomly assigned in a ratio of *** among the *** treatment groups described in Figure 1.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
Figure 1. Schematic Representation of Study Design.
Approximately 1250 subjects will be randomized *** at up to *** study sites. The randomization schedule will be stratified by *** to ***; at least *** of subjects enrolled into the study will be ***.
Subjects will initiate treatment with a dose titration during weeks 1-4 with doses gradually increased at *** intervals, as determined by randomization assignment, until the specified dose is reached and will be treated for 52 weeks (weeks 5-56) at the assigned dose level. Subjects will return to the site at the end of weeks 2 and 4 (Visits 3 and 4) during titration and at 4-week intervals thereafter. A schedule of events is provided in Appendix 1.
The primary endpoints are differences between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), and percent of subjects with weight loss of 5% or more at end of treatment (week 56). Percent weight loss will be calculated as ***.
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) for *** and *** groups.
· The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups.
· The difference in change in waist circumference (randomization to week 56) for *** and *** groups.
Additional efficacy endpoints will include the effect of treatment on obesity-associated cardiovascular risk factors (total cholesterol, triglycerides, LDL-C, HDL-C, fasting glucose, blood pressure), ***, *** of *** and *** and ***, *** and ***. The difference between *** and *** groups in the rate of progression to type 2 diabetes will be calculated. Baseline adjusted Framingham 10-year risk scores will be calculated and compared between groups at weeks 28 and 56. Change in BMI between baseline and week 28 and end of treatment (Visit 17, week 56) will be evaluated. Additionally, body composition will be assessed by *** at ***. These evaluations will be made at ***, and at *** and ***.
The change in weight loss (percent, absolute, percent of subjects achieving weight loss of > 5% and > 10% of starting weight), waist circumference and obesity associated risk factors will also be assessed over time. Subgroup analyses, including but not limited to analysis by gender, age and race, may be performed.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Safety evaluations will include assessment of adverse events, including eye symptoms, ***.
*** will also be obtained, and effects of various cofactors including (but not limited to) ***, gender, race, ***, and age will be evaluated. ***.
4 SUBJECT SELECTION
This clinical trial can fulfill its objectives only if appropriate subjects are enrolled. The following eligibility criteria are designed to select subjects for whom protocol treatment is considered appropriate. All relevant medical and non-medical conditions should be taken into consideration when deciding whether this protocol is suitable for a particular subject.
4.1 Inclusion Criteria
This study is designed to treat obese adult subjects with BMI > *** who have *** or whose *** using reasonable measures. To be eligible for enrollment into this trial, subjects must meet all of the following criteria. Specifically, subjects must:
1. Be adults 70 years of age or less;
2. Have a BMI > ***;
3. If females of child-bearing potential, be using adequate contraception, defined as double barrier methods, stable hormonal contraception plus single barrier method, or tubal ligation. Females are considered to be of child-bearing potential unless they have undergone a hysterectomy or bilateral oophorectomy, are 55 years of age or greater and have experienced spontaneous cessation of menses for at least 1 year, or have a documented follicle stimulating hormone level >40 IU/L;
4. Have a ***;
5. Have blood pressure of ***;
6. Have a fasting blood glucose level of ***;
7. Provide written informed consent; and
8. Be willing and able to comply with scheduled study visits, treatment plan, laboratory tests and other study procedures.
4.2 Exclusion Criteria
Subjects presenting with any of the following will not be included in the trial:
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
1. Known allergy or hypersensitivity to phentermine or topiramate, any prior use of a combination of phentermine and topiramate for weight loss or use of phentermine or topiramate for any indication within the past 3 months;
2. Weight gain or loss of greater than 5 kg, use of a very low-calorie diet, or participation in a formal weight loss program (investigational or otherwise) within the past 3 months (this includes: Weight Watchers and related dietary/lifestyle intervention programs; prepared food programs; prescribed or over-the-counter weight loss medications; dietary supplement or herbal preparations, teas, or tinctures intended for weight loss; or any supervised fast or very low calorie diet);
3. Obesity that is of a known genetic or endocrine origin;
4. History of any eating disorders (e.g. bulimia, binge eating disorder) within the past year;
5. History of drug abuse within the past year;
6. History of alcohol abuse (defined as >14 drinks per week) within the past year;
7. Previous bariatric surgery;
8. Diagnosis of type 2 diabetes by history, or as confirmed by a fasting blood glucose of 126 mg/dL or greater, or history of any antidiabetic medication use;
9. Smoking cessation within the previous 3 months or plans to quit smoking during study participation;
10. History of glaucoma, history of increased intraocular pressure or any past or present use of medications to treat increased intraocular pressure;
11. Clinically significant thyroid dysfunction as evidenced by signs or symptoms of hypothyroidism, a TSH > 1.5 x ULN, or use of thyroid hormone treatment that has not been stable for at least 3 months;
12. Use of chronic systemic glucocorticoid therapy, or any other steroid hormone therapy that has not been stable for at least 3 months;
13. Any history of bipolar disorder or psychosis, greater than one lifetime episode of major depression, current depression of moderate or greater severity (PHQ-9 score of 10 or more), presence or history of suicidal behavior or ideation with some intent to act on it, or antidepressant use that has not been stable for at least 3 months;
14. Stroke, myocardial infarction, life-threatening arrhythmia or coronary re-vascularization within the past 6 months;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
15. Unstable angina, congestive heart failure (NYHA Class II, III or IV), or known or suspected clinically significant cardiac valvulopathy;
16. Blood pressure medication that has not been stable for at least 1 month;
17. Any history of malignancy within the past 5 years other than basal or squamous cell carcinoma of the skin that has been completely excised or cervical cancer that has been surgically excised.
18. Cholelithiasis within the past 6 months;
19. Any history of nephrolithiasis;
20. For female subjects of childbearing potential, pregnancy, breastfeeding, or plans for pregnancy during the study period;
21. Use of any investigational medication or device for any indication within the last month;
22. Evidence of any clinically significant renal, pulmonary, hepatic, psychiatric or other condition by history, physical examination or laboratory studies that, in the opinion of the investigator, would contraindicate the administration of study medications, affect compliance, interfere with study evaluations or confound the interpretation of study results.
4.3 Randomization Criteria
The following criteria must be met prior to randomization and dispensing of study medication to subjects:
1. Baseline physical examination, ECG and laboratory findings with no abnormalities that are considered clinically significant by the principal investigator;
2. Laboratory values that are within the ranges specified below:
***
4.4 Life Style Guidelines
Subjects randomized into the study will receive counseling on how to reduce their caloric intake by 500 kcal/day, will be advised to increase their daily water intake and will be advised to implement a simple exercise program, as tolerated. Subjects will be advised to initiate a lifestyle change program utilizing the LEARN® Program for Weight Management.(20) The LEARN® program is a 16-week program designed to aid in weight management by providing tools to facilitate lifestyle, attitude, relationship, nutrition and exercise changes. Each subject will be
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
provided with a LEARN® manual and advised to read and implement the material as appropriate to their individual situation. Site personnel will be encouraged to discuss these materials with subjects at their regularly scheduled visits. However, no data will be collected to document the level of compliance with the program’s dietary, lifestyle and/or exercise recommendations.
To facilitate discussions between study subjects and research staff, subjects will complete a 24-hour dietary recall at their randomization visit (Visit 2); however, information provided as part of this exercise will be used only for discussions related to recommended dietary modification and will not be retained as documentation. No data will be collected during the study to document the level of compliance with dietary recommendations.
5 TRIAL TREATMENTS
5.1 Allocation to Treatment
Subjects meeting the randomization requirements specified in Section 4.3 will be randomized to study treatment at Visit 2 using a centralized computer-generated randomization system. Eligible subjects will be randomly assigned to *** or *** at a ratio of ***. Randomization will be stratified by ***, and at least *** of subjects will be ***. Both the subject and the study site will be blinded as to subject randomization.
To implement subject randomization among treatment groups, each participating site will be pre-stocked with titration kits corresponding to each treatment group. When a subject qualifies for randomization, study site personnel will contact an Interactive Voice Response System (IVRS), either by telephone or through a designated web site, and provide the information required regarding the study subject. The randomization assignment will then be made, and the site will be instructed to dispense a specific kit number to the study subject. Additional kits will be shipped to the site to replace those dispensed according to the randomization schedule. When subjects return for subsequent study visits, the IVRS system will be used to dispense replacement study medication kits, as appropriate.
5.2 Breaking the Blind
Study medication must not be unblinded during the study unless it is considered absolutely necessary by the investigator for the management of an adverse event or other medical emergency. Under such conditions, the identity of the study treatment will be obtained by contacting the IVRS. Any subject who is unblinded during the study will be discontinued from participation in the trial.
VIVUS, Inc. will be notified of any unblinding of subject treatment group ***. The investigators are also required to ensure that any potential serious adverse events are reported according to the requirements outlined in Sections 8.2 and 8.5 and to provide a written report to document the reason for unblinding within ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
5.3 Drug Supplies
5.3.1 Formulation and Packaging
Medications for this trial will consist of ***. Doses specified for each treatment group will be achieved by varying the *** added to each capsule. Regardless of the dosage assignment, all study treatments will be administered as a ***.
Clinical supplies will be manufactured for VIVUS, Inc. by *** in accordance with current Good Manufacturing Practices. All clinical supplies will be labeled with information required by national and/or international regulations. Study drug will be packaged into 2 types of kits; titration kits for use during the first 4 weeks of study therapy when doses are being increased gradually to the final assigned dose, and treatment kits, for use once subjects have been titrated to their assigned dosage of medication. Each titration kit contains *** for use during weeks 1 through 4 of titration, with each ***. Each *** on the *** will be labeled with the *** and will contain capsules with the dose specified for that week of treatment, as outlined in Table 1.
Table 1: VI-0521 Dosage Strengths by Titration Week for Each Treatment Group
|
|
|
*** |
|
*** |
| ||||||
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
Titration kits will consist of *** labeled with the ***. Each *** will be labeled with the ***.
Treatment kits will consist of bottles, *** of study medication at the treatment dosages (week 4) shown in Table 1. Each kit will contain a single bottle, will be labeled with the ***.
5.3.2 Preparation and Dispensing
Clinical supplies provided by the sponsor are to be dispensed only by or under the direct supervision of qualified investigators to subjects meeting the criteria for study entry and in accordance with this protocol. Assignment of specific titration and treatment drug kits to study subjects will require the use of the IVRS system; however, no other preparation of clinical supplies is required of the investigational staff.
5.3.3 Administration
Investigators will instruct subjects to take 1 capsule of study medication every morning. When dispensing titration kits, investigators should ensure that subjects understand that each card contains a 4-week supply of medication, and that the capsules must be taken ***. Investigators will also instruct subjects to return the study medication kit to the site at each site visit. For the
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
week 2 visit, the site will perform drug accountability and return the *** to the subject for use during titration weeks 3 and 4.
5.3.4 ***
*** is an option for subjects who experience ***. *** is implemented through the ***, and will be done without ***. *** is not an option for subjects who experience ***.
When *** is not appropriate or when *** may be required due to events unrelated to subject treatment, subjects may ***. *** are possible with agreement from the medical monitor. All subjects undergoing *** for *** may be *** based on discretion of the PI. If dosing has been ***, a new titration kit should be ordered through *** to ***. Subjects who have a ***. For subjects having a ***. For ***, subjects may ***.
If ***, subjects may be ***. Subjects *** will be encouraged to *** and to continue to make site visits at the regularly scheduled intervals. The last date on which the subject ***. If the subject ***. If the subject elects to withdraw completely from the trial (Section 6.4), the end of treatment (Visit 17) testing should be completed.
5.3.5 Compliance
Subject compliance with study medication will be assessed by ***, and *** should implement any corrective action necessary. Subjects who remain noncompliant with study dosing despite corrective actions by site personnel may be discontinued from the trial.
5.4 Drug Storage and Drug Accountability
All unused study drug must be stored in its packaging at room temperature in a dry, secure area. Access to drug storage areas should be limited to the investigator and designated staff involved with the study. All used and unused drug must be maintained at the study site and made available for audits by VIVUS, Inc. personnel or their designee. It should be noted that one component of the study drug combination is a Schedule IV controlled substance. The investigator should take all appropriate measures to control access to and dispensing of study drug.
The investigator must maintain records documenting the amount, condition and date of delivery of all study drug received from the sponsor. In addition, all drug dispensed to study subjects during the course of the study must be ***. Subjects must be instructed to *** by each subject. No investigational drug or packaging, used or unused, may be discarded. All packaging and used and unused drug must be returned to the sponsor or designated representative upon completion of the study.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
5.5 Concomitant Medication(s)
5.5.1 Excluded Medications
Subjects must not take the following medications during their participation in this trial:
· ***;
· ***;
· ***;
· ***;
· ***.
Although the need for any antidiabetic medications at screening would exclude subjects from trial participation, subjects who develop a need for these medications during the course of the trial need not be discontinued for this reason alone. Refer to Section 5.6 for treatment and monitoring recommendations.
5.5.2 Other Restricted Medications
Subjects using *** or allowed *** must be on doses that have been stable for at least 3 months prior to screening. For subjects who develop symptoms consistent with *** during the study, *** replacement may be initiated following appropriate diagnostic work-up (Section 5.8). In the event that subjects require any other changes in these medications, the sponsor should be contacted regarding their continued eligibility.
*** and *** are permitted, provided that the dosage has been stable for at least 1 month prior to screening, and the frequency of use does not exceed twice a week.
All other medications used for the treatment of *** associated with *** must be stable for at least 1 month prior to screening. However, adjustment of these medications during the trial is permitted if the subject’s requirements for treatment change.
5.5.3 Documentation of Concomitant Medication Use
All concomitant medications, including both prescription and over-the-counter products, vitamins and nutritional/herbal supplements, must be listed on the appropriate case report form at trial entry. Any change in concomitant medication during the course of the trial must be noted on the appropriate CRF.
5.6 Treatment of Diabetes
Subjects who become diabetic during the course of the study will be provided with ***. They will be instructed to ***. Diabetic subjects will ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Subjects who develop elevated fasting blood glucose values during the study should ***. *** is suggested as the initial therapy for *** type 2 diabetes ***. ***, including ***, should be reserved for subjects who cannot achieve adequate control with other modes of treatment. *** are prohibited, and subjects requiring treatment with these medications must be discontinued from the trial. Subjects whose blood glucose cannot be adequately controlled with the concomitant treatments allowed in this trial should be maintained in the study but discontinued from treatment, and referred back to their primary health care provider for more intensive treatment (see Section 6.4).
During treatment, subjects whose fasting blood glucose is ***.
5.7 Treatment of ***
For subjects whose ***. If these medications are already present, ***.
Subjects whose ***, should be discontinued from study treatment and referred back to their primary healthcare provider for more intensive management. Subjects may continue attending study visits ***.
Subjects whose ***.
5.8 Treatment of ***
Individuals who experience rapid weight loss sometimes ***. Subjects who develop symptoms of *** need not be discontinued from therapy. These subjects should be assessed clinically and with appropriate laboratory testing ***. Subjects found to be *** may be ***, as appropriate.
6 TRIAL PROCEDURES
This trial is being conducted in conjunction with a companion study (OB-303: A Phase III Randomized, Double-Blind, Placebo Controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in Adults with Obesity-Related Co-Morbid Conditions) that is of similar design. The distinguishing factors between the two trials are related to the subject populations, specifically, the subjects enrolled into this trial will have a *** associated with obesity than the subjects enrolled into study OB-303. Because there is little overlap between the populations for these two trials, the majority of sites participating in this trial will also be conducting study OB-303. Because results of the screening evaluations may be required to determine which trial a particular subject qualifies for, the initial screening procedures for both of these trials are identical, and will be done under a generic screening informed consent. Once the determination of trial placement has been made a specific informed consent document for that study will be obtained and specific trial evaluations will continue. A schedule of study activities by visit is presented in Appendix 1. A detailed list of these activities is provided in the following sections.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
6.1 Screening (Visit 1)
Activities at screening (Visit 1) are:
· Obtain written informed consent for screening evaluations;
· Obtain demographics (including age, gender, race, ethnicity, education, history of smoking) and medical history (including history of hypertension, vascular disease and dyslipidemia) and family history of hypertension, smoking and premature cardiovascular disease
· Assess weight (kg), height (cm), waist circumference (cm) and BMI (note: the BMI calculation must be obtained from the IVRS following informed consent and should be obtained prior to initiating the testing described below);
· Assess ▇▇▇▇▇ ▇▇▇▇▇;
· Record baseline concomitant medications;
· Administer *** and ***;
· Administer ***;
· Assess Inclusion/Exclusion criteria;
· Obtain blood and urine samples for laboratory testing (including TSH);
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
· Schedule the randomization visit in 2 weeks (± 7 days).
6.2 Treatment Period
6.2.1 Randomization (Visit 2)
Subjects eligible for treatment will be randomized and study drug dispensed at Visit 2. Activities at randomization are:
· Obtain study-specific written informed consent;
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
· Obtain blood sample for laboratory testing (biomarkers);
· Perform complete physical examination, including neurological examination and auscultation for heart sounds (may be performed between Visits 1 and 2 after results from Visit 1 indicate subject may be eligible to participate);
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Obtain written informed consent for *** procedures and perform *** scan ***. *** scans may be performed between Visit 1 and Visit 2, after results from Visit 1 indicate subject may be eligible to participate;
· Perform 12-lead electrocardiogram (may be performed between Visits 1 and 2 after results from Visit 1 indicate subject may be eligible to participate);
· Assess adverse events (including eye symptoms), if any;
· Obtain ▇▇▇▇▇ ▇▇▇▇▇;
· Obtain weight and waist circumference measurements;
· Assess changes in concomitant medications;
· Administer *** for *** and ***;
· Evaluate randomization criteria (Section 4.3);
· If subject is eligible, contact IVRS and obtain subject study medication identification;
· Obtain 5 mL serum sample to be retained for possible use for scientific exploratory testing;
· Complete 24 hour dietary recall, distribute LEARN® materials and perform lifestyle counseling;
· Dispense study medication *** from the assigned titration kit and provide instructions for proper use;
· Schedule the next study visit in 2 weeks (± 2 days).
6.2.2 Titration (Visit 3)
Subjects will visit the study site at the end of the second week of titration (Visit 3). Activities at Visit 3 are:
· Obtain weight and waist circumference measurements;
· Obtain ▇▇▇▇▇ ▇▇▇▇▇;
· Assess adverse events (including eye symptoms), if any;
· Assess concomitant medications;
· Administer *** and ***;
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Review the study medication used during weeks 1 and 2; assess treatment compliance and perform drug accountability. Return the titration card to the subject for use during weeks 3 and 4;
· Perform brief review of lifestyle counseling and answer questions, if any;
· Schedule the next study visit in 2 weeks (± 2 days).
6.2.3 Treatment (Visits 4 through 16)
Subject titration will be completed on Visit 4; subsequently, subjects will return to the site at 4-week intervals for evaluation and to obtain additional study medication. Activities will be as follows:
· Obtain weight and waist circumference measurements;
· Obtain ▇▇▇▇▇ ▇▇▇▇▇;
· Assess adverse events (including eye symptoms), if any;
· Assess concomitant medications;
· Administer *** and ***;
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
· Collect study medication from previous visit; assess for treatment compliance and perform drug accountability;
· Perform brief review of lifestyle counseling and answer questions, if any;
· Obtain blood samples for blood chemistry testing (Visits 4, 5, 7 and 10 only);
· Administer *** for *** and *** (Visits 4, 7 and 10 only);
· Ensure that the previous dose of trial medications was taken between 20 and 28 hours ago, and obtain a *** blood sample for *** evaluations (if the previous dose of trial medications is outside this window, *** should be postponed for 1 day) (Visits 7 and 10 only);
· Obtain blood samples for hematology testing (Visit 10 only);
· Administer *** (Visit 10 only);
· Perform *** scan *** (Visit 10 only);
· Dispense study medication for the upcoming study interval and provide the proper instructions for use;
· Schedule the next study visit in 4 weeks (± 1 week).
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
6.2.4 End of Treatment (Visit 17 or withdrawal from study)
The end of treatment for subjects completing the study is Visit 17 (week 56). End of treatment testing will also be performed for subjects who are withdrawn from the study prior to completion of the study at the time of their treatment termination. For subjects who withdraw from the study prior to completion, the site will also attempt to contact the subject at or about the 56 week time point to obtain end of study assessments (except ***).
Activities at the end of treatment visit include:
· Obtain weight and waist circumference measurements;
· Obtain ▇▇▇▇▇ ▇▇▇▇▇;
· Assess adverse events (including eye symptoms), if any;
· Assess concomitant medications;
· Administer ***;
· Administer ***;
· Administer *** for *** and ***;
· Administer ***;
· Complete End of Treatment Questions: ***;
· Perform *** scan ***;
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
· Obtain blood and urine samples for laboratory testing;
· Perform complete physical examination, including neurological examination and auscultation for heart sounds;
· Perform 12-lead electrocardiogram;
· Collect study medications dispensed at previous visit, assess treatment compliance and perform drug accountability.
6.3 Study Period
The study period for each subject will begin when written informed consent is provided and will continue until Visit 17 (week 56 or early termination) is completed. Sites should link the scheduling of visits to the randomization visit (Visit 2). Visit windows are provided to allow subject and site scheduling convenience. However, every effort should be made to ensure that visits occur within these windows so that the overall treatment duration is 56 weeks, including
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
titration period, for subjects who complete all visits. In certain instances, adverse event information may be required for events occurring after the study period (Section 8.4).
6.4 Subject Withdrawal
Subjects may withdraw from the trial at any time and for any reason. Additionally, the subject may be withdrawn because of:
· Adverse event;
· Subject lost to follow-up;
· Requirement for other medical treatment excluded by the protocol;
· Lack of compliance with the provisions of the protocol;
· Treatment unblinded by investigator;
· Pregnancy;
· Lack of efficacy;
· Termination of trial.
Withdrawn subjects will not be replaced.
Subjects discontinued from treatment should be encouraged to remain in the trial off-treatment and to continue site visits at the scheduled intervals (Section 5.3.4). Subjects who withdraw completely from the trial at any point should complete the end of treatment (Visit 17, week 56) testing. The date of last dose should be recorded.
Every effort should be made to document subject outcome. For subjects who elect to withdraw from the trial without continuing site visits, the investigator should ***.
At about the 56 week time point, withdrawn subjects who have not continued site visits should be asked to return to the site to obtain weight and waist circumference measurements at a minimum and, if possible, the following additional information: adverse events, ▇▇▇▇▇ ▇▇▇▇▇, laboratory tests (chemistry, hematology, urinalysis), concomitant medications, questionnaires ***.
If a subject withdraws from the trial and also withdraws consent for disclosure of future information, ***. ***.
Investigators must discontinue trial participation for all subjects if the sponsor terminates the trial, and evaluations that would normally be performed upon trial completion should be made at that time. For subjects who have received ***, this includes obtaining information requested for the Visit 17 (week 56) end of treatment visit.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
7 ASSESSMENTS
7.1 Weight, Waist Circumference, Height and BMI Assessment
Subjects will be weighed (kg) and a waist circumference (cm) measurement obtained at each study visit (Visits 1-17). Height (cm) and BMI will be determined at the screening visit (Visit 1) only.
7.1.1 Weight Assessment
Subjects should be weighed in kilograms using a calibrated digital scale. The same scale should be used for each measurement and measurements should be evaluated by the same site personnel at each visit, whenever possible. Subject weights should be obtained, whenever possible, under the same conditions (no shoes, clothing of similar weight) that were employed at the first (screening) weighing. Subjects should be encouraged to complete their weigh-in visits in the morning and should be fasting prior to weigh-in.
7.1.2 Waist Circumference Measurement
Waist circumference measurements (cm) will be performed using a measuring tape provided by VIVUS, Inc., and should be obtained by the same individual at each visit, when possible. To measure the waist circumference, locate the top of the right iliac crest. Place the measuring tape in a horizontal plane (parallel to the floor) around the abdomen at the level of the top of the iliac crest as shown in Figure 2.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
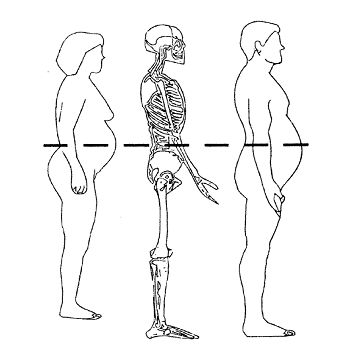
Figure 2. Measuring Tape Position for Waist Circumference Assessments
Ensure that the subject is relaxed. Ensure that the tape is snug but does not indent or compress the skin, and make the measurement at the end of a normal expiration.(21)
7.1.3 Height and BMI
Height measurements (cm) and BMI (kg/m2) will be determined by the site at screening only. Height measurements should be made without shoes.
BMI will be calculated for purposes of screening using the IVRS. A printed copy of the BMI obtained from the IVRS should be retained as part of the subject’s source documentation. Manual BMI calculations should not be performed. If a subject does not meet the BMI criterion for inclusion into the study, no further screening procedures should be undertaken.
7.2 ▇▇▇▇▇ ▇▇▇▇▇
▇▇▇▇▇ ▇▇▇▇▇ (systolic blood pressure, diastolic blood pressure, heart rate, respiration rate, temperature) will be assessed at each study visit. Subjects should be seated comfortably for at least *** prior to assessing ▇▇▇▇▇ ▇▇▇▇▇. Pulse rate and respiratory rate measurements should be made by counting events (heartbeats or breaths) for a period of 30 seconds and multiplying these values by 2 to obtain the rates per minute. A calibrated cuff should be employed for blood pressure measurements. Whenever possible, the same person should perform all assessments for a given subject.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
7.3 Questionnaires
7.3.1 ***
The *** for the assessment of ***.(22)-(24)
Because this instrument is intended to be completed ***, it is important that *** on this questionnaire in any way. Should subjects ***. Because this questionnaire assesses the ***. Site personnel, therefore, must carefully review questionnaires for completeness before ***, and assure that questionnaires are properly completed.
The *** is being used ***. This questionnaire will be completed at ***, and at *** after the ***. Answers to the questionnaire may reveal evidence of ***, including the ***. It is the responsibility of the investigator to evaluate ***. The evaluation by the Investigator will be guided by the ***. Investigators should document any such problems ***. It is expected that any randomized ***.
7.3.2 ***
The ***(25) is an ***. Each of the ***, and is answered on a yes/no basis. This assessment will be administered to all *** at *** in order to confirm the *** included in the treatment program. Subsequent *** evaluations will be done at *** after ***. All *** assessments must be administered by a trained interviewer. If any assessments reveal ***, then the results must be reviewed by a physician investigator prior to ***.
7.3.3 ***
The *** that will be completed at *** and ***.
This questionnaire is designed to evaluate the ***.(26), (27) This instrument is intended to be completed ***.
Site personnel must ***. It is critical, therefore, that site personnel review questionnaires for completeness at the time they are initially filled out, and that any missing answers are completed before the ***.
7.4 *** and End of Treatment Questions
7.4.1 ***
*** for *** and *** will be assessed at *** and at the ***. For the *** assessment, subjects will *** For the *** assessment, a ***
7.4.2 End of Treatment Questions
At the end of treatment, subjects will be asked to respond to *** questions assessing ***. These questions are:
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· ***.
· ***.
· ***
7.5 Laboratory Tests
Laboratory tests will be performed at a licensed, certified central testing laboratory identified by the sponsor. Urine pregnancy tests will be performed at the site using kits supplied by VIVUS, Inc. Testing will be conducted according to the schedule provided below. For blood chemistry tests, the subject must have fasted for a minimum of 8 hours before the sample can be drawn.
Testing for phentermine and topiramate will be conducted in a sponsor-approved central laboratory using validated laboratory assays.
7.5.1 Blood Chemistry
Fasting blood chemistries will be evaluated at screening (Visit 1), Visit 4 (week 4), Visit 5 (week 8), Visit 7 (week 16), Visit 10 (week 28) and end of treatment (Visit 17 or study withdrawal). Tests will be made for the following: albumin, alkaline phosphatase, alanine transaminase (ALT), aspartate amino transferase (AST), blood urea nitrogen (BUN), serum calcium, serum chloride, serum sodium, bicarbonate, creatinine, creatinine clearance, direct bilirubin, gamma-glutamyl transferase (GGT), glucose, lactate dehydrogenase (LDH), serum phosphorus, serum potassium, total bilirubin, total cholesterol, total protein, uric acid, triglycerides, HDL cholesterol (HDL-C) and LDL cholesterol (LDL-C).
7.5.2 Hematology
Hematology studies will be evaluated at screening (Visit 1), Visit 10 (week 28) and end of treatment (Visit 17 or study withdrawal) for hemoglobin, hematocrit, red blood cell count (RBC), total white blood cell count (WBC), WBC differential (neutrophils, lymphocytes, monocytes, eosinophils, basophils) and platelet count.
7.5.3 Urinalysis
A routine midstream urinalysis with reflex microscopic evaluation will be obtained at screening (Visit 1) and end of treatment (Visit 17 or study withdrawal).
7.5.4 Biomarkers
Blood biomarkers including C-reactive protein, adiponectin, and fibrinogen will be evaluated at baseline (Visit 2) and end of treatment (Visit 17 or study withdrawal). All post-screening biomarker results will be blinded to investigators and sponsor.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
7.5.5 Urine Pregnancy Test
A urine pregnancy test will be obtained for females of childbearing potential at each study visit (Visits 1-17). Urine pregnancy testing will be performed at the site and results reported on the appropriate CRF.
Female subjects who have undergone a hysterectomy or bilateral oophorectomy, who have a follicle stimulating hormone level > 40 IU/L or who are 55 years of age or greater and have experienced cessation of menses for at least 1 year are considered to not be of childbearing potential. Urine pregnancy testing is not required in these subjects.
Management of any subject who becomes pregnant during this study is described in Section 8.5.3.
7.5.6 Thyroid Stimulating Hormone (TSH)
A test for thyroid stimulating hormone (TSH) will be conducted at screening (Visit 1).
7.5.7 Antibody to HIV, HCV, HBsAg
Testing for hepatitis B surface antigen (HBsAg) and antibodies to human immunodeficiency virus (HIV) and hepatitis C virus (HCV) will be conducted at screening.
7.5.8 Urine Drug Screen
A urine drug screen will be conducted at screening. Subjects will be tested for cannabinoids, cocaine, amphetamines, opiates and phencyclidine.
7.5.9 Screening Serum Sample (Retain)
A 5.0 mL serum sample will be obtained at Visit 2 to be retained at the central laboratory for possible use in scientific experimental studies.
7.6 *** Evaluation
Samples for the evaluation of *** will be obtained at ***.
Subjects should be advised at the previous visit to refrain from dosing with study medication until after the clinic visit on *** and should be reminded about 1 week prior to the visit to delay dosing on the days of *** until after testing at the site. If the subject has taken study medication < 20 hours prior to the clinic visit, *** should be postponed until the next day.
7.7 Physical Examination
A complete physical examination will be performed prior to randomization (between Visits 1 and 2 or at visit 2) and at Visit 17 (week 56) or early withdrawal. The screening physical
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
examination should not be completed until results of testing at Visit 1 indicate that the subject may be eligible for entry into the trial.
The physical examination will consist of the following: general appearance, skin, head, eyes, ears, nose, throat, neck (including thyroid), lymph nodes, chest, heart (including auscultation for heart sounds and murmurs), abdomen, extremities, and neurological examination. Subjects will be evaluated for clinically significant abnormalities that would prevent entry into the study and for clinically significant differences between screening and end of treatment.
7.8 Electrocardiogram (ECG)
Twelve-lead electrocardiographic studies will be obtained prior to randomization (between Visits 1 and 2 or at Visit 2) and the end of treatment (Visit 17 or early withdrawal). Screening ECG studies should not be performed until results of testing at Visit 1 have been reviewed to confirm that the subject may be eligible for treatment. Whenever possible, ECGs should be obtained in the morning with the timing of the studies matched as closely as possible.
Studies will be evaluated for clinically significant abnormalities that would prevent entry into the study and for clinically relevant changes between screening and end of treatment. Parameters including R-R, QRS, QT, and QTc intervals will also be recorded.
7.9 Framingham Risk Score
The Framingham risk assessment evaluates the 10-year risk for development of coronary heart disease.(28) In order to calculate the Framingham risk, the following information will be recorded at screening: demographics (age, gender, race, and ethnicity), medical history (including histories of smoking, diabetes, congestive heart failure, myocardial infarction, hypertension, and dyslipidemia) and family history of premature cardiovascular disease. The Framingham risk assessment will be calculated by VIVUS, Inc. or its designee based on data provided on the CRF.
7.10 Body Composition
At a selected subset of study sites, body composition assessments will be made at baseline (Visit 2), week 28 (Visit 10) and week 56 (Visit 17) using ***. Scans will be collected at a sufficient number of study sites to provide body composition data on approximately *** subjects from this study combined with protocol OB-303. Equipment and procedures used to obtain *** data will be standardized as described in a separate document. All sites will be trained on these procedures prior to performing scans on study subjects. Scans will be read at a central facility and the reader will be blinded.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
8 ADVERSE EVENTS
8.1 Adverse Events
Adverse events (AEs) are defined as any untoward medical occurrences in subjects administered the trial treatment, whether or not they have a causal relationship to the treatment. All observed or volunteered adverse events regardless of suspected causal relationship to the investigational product must be reported as described in the following sections.
The investigator must pursue and obtain information adequate to describe adverse events, their severity and relationship to study treatment, and their outcomes. Descriptions of neurological or psychological adverse events should be consistent with standard diagnostic criteria and terminology (such as DSM-IV) rather than general reports of symptoms. For adverse events with a causal relationship to the investigational product, follow-up by the investigator is required until the events or their sequelae resolve or stabilize at a level acceptable to the investigator, and VIVUS, Inc. concurs with that assessment. Investigators must also assess whether adverse events meet the criteria for classification as serious adverse events (see Section 8.2) requiring immediate notification to VIVUS, Inc. or its designated representative.
8.1.1 Severity Assessment
The investigator will assess the severity of all adverse events using the ***, ***, or *** to describe the maximum intensity of each adverse event. For purposes of consistency, these intensity grades are defined as follows:
· ***;
· ***;
· ***.
Note the distinction between the severity and the seriousness of an adverse event. A *** is not necessarily a ***. For example, a headache may be *** but would not be classified as serious unless it met one of the criteria for ***.
8.1.2 Causality Assessment
Trial investigators are required to provide an assessment of causality for all adverse events (serious and non-serious) observed during this trial. This assessment will provide a determination of whether, in the investigator’s judgment, there exists a reasonable possibility that the investigational product caused or contributed to an adverse event. For this assessment, investigators must categorize the causality as either “related” or “not related.” For an adverse event to be considered “related” to the trial treatment, there should be evidence that the event follows a reasonable temporal sequence from the administration of trial treatment or that the event follows a known response pattern to the drug. Causality would be further confirmed by
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
improvement in the adverse event upon stopping the trial treatment and reappearance of the event upon rechallenge.
8.1.3 Abnormal Test Findings
The criteria for determining whether an abnormal objective test finding should be reported as an adverse event are as follows:
· Test result is associated with accompanying symptoms, and/or
· Test result requires additional diagnostic testing or medical/surgical intervention, and/or
· Test result leads to a change in trial dosing or discontinuation from the trial, significant additional concomitant drug treatment, or other therapy, and/or
· Test result is considered by the investigator or sponsor to represent a clinically significant finding.
Merely repeating an abnormal test, in the absence of any of the above conditions, does not constitute an adverse event. Any abnormal test result that is determined to be an error does not require reporting as an adverse event.
8.2 Serious Adverse Events
As defined in the Code of Federal Regulations (21 CFR 312.32), a serious adverse event (SAE) or serious adverse drug reaction is any untoward medical occurrence at any dose that:
· Results in death;
· Is life-threatening (immediate risk of death);
· Requires inpatient hospitalization or prolongation of existing hospitalization;
· Results in persistent or significant disability/incapacity;
· Results in congenital anomaly/birth defect.
Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered serious adverse drug experiences when, based on appropriate medical judgment, they may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes listed above. Adverse events that, in the investigator’s judgment, significantly jeopardize trial subjects or require medical or surgical intervention in order to prevent any of the outcomes listed above should therefore be reported as serious adverse events.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
8.2.1 Definition of Hospitalization
Adverse events reported from clinical trials that result in hospitalization or prolong an existing hospitalization are considered serious. Any initial admission (even if less than 24 hours) to a healthcare facility meets these criteria.
Outpatient ambulatory surgical procedures (same day surgeries) and routine emergency room treatment do not qualify as hospitalizations. Additionally, hospitalization in the absence of a precipitating, clinical adverse event is not in itself a serious adverse event. Examples include, but are not limited to:
· Admission for treatment of a preexisting condition not associated with the development of a new adverse event or with a worsening of the preexisting condition (e.g., for work-up of persistent pre-treatment lab abnormality);
· Administrative admission (e.g., for yearly physical exam);
· Optional admission not associated with a precipitating clinical adverse event (e.g., for elective cosmetic surgery);
· Pre-planned treatments or surgical procedures should be noted in the baseline documentation for the entire protocol and/or for the individual subject.
Diagnostic and therapeutic non-invasive and invasive procedures, such as surgery, should not be reported as adverse events. However, the medical condition for which the procedure was performed should be reported if it meets the definition of an adverse event. For example, an acute appendicitis that begins during the adverse event reporting period should be reported as the adverse event, and the resulting appendectomy should be recorded as treatment of the adverse event.
8.3 Eliciting Adverse Event Information
The investigator is to report all directly observed adverse events and all adverse events spontaneously reported by the trial subject. In addition, trial subjects should be ***
Certain adverse events require prompt and specific action by the investigator in any clinical trial. The following sections describe additional requirements to ensure ***.
8.3.1 Eye Pain
At each visit, subjects will be queried regarding eye symptoms, *** Subject responses will be recorded as adverse events, where appropriate. If any subject reports eye pain and/or ***, the subject should be referred to *** or ***. Treatment with study drug should be discontinued until the ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
8.3.2 ***
All subjects will be screened for the presence and *** at *** and subsequently at *** after the *** using a validated survey instrument *** designed for assessment of *** in a primary care setting. The *** is a *** module based directly on the diagnostic criteria for ***. *** will also be assessed at ***and at *** following the *** using the ***.
Should this additional assessment indicate the presence ***. Any such event must be ***. Subjects must be ***.
Any *** must be ***.
8.4 Reporting Period
The reporting period for adverse events begins when the subject provides written informed consent and extends until *** after the last dose of the investigational product is administered. All adverse events that occur during this period and are known to the investigator must be reported according to the requirements outlined in Section 8.5.
8.5 Reporting Requirements
All adverse events will be reported on the adverse event page(s) of the CRF. In addition, serious adverse events must also be reported on a separate serious adverse event form. Where the same data are collected on both AE and SAE forms, the forms must be completed in a consistent manner. For example, the same adverse event term should be used on both forms. Adverse events should be reported using concise medical terminology on the CRFs as well as on the form for collection of serious adverse event information.
8.5.1 Serious Adverse Event Reporting Requirements
If a serious adverse event occurs, VIVUS, Inc., or designee, is to be notified within 1 business day of awareness of the event by the investigator. In particular, if the serious adverse event is fatal or life-threatening, notification to VIVUS, Inc., or designee, must be made immediately, irrespective of the extent of available adverse event information. This timeframe also applies to additional new information (follow-up) on previously forwarded serious adverse event reports.
***
For all serious adverse events, the investigator is obligated to pursue and provide information to VIVUS, Inc. in accordance with the timeframes for reporting specified above. In addition, an investigator may be requested by VIVUS, Inc. or designee to obtain specific additional follow-up information in an expedited fashion. This information may be more detailed than that captured on the AE or SAE CRFs. In general, this will include a description of the adverse event in sufficient detail to allow for a complete medical assessment of the case and independent determination of possible causality. Information on other possible causes of the event, such as
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
concomitant medications and illnesses, must be provided. In the case of a subject death, a summary of available autopsy findings must be submitted as soon as possible to VIVUS, Inc. or its designated representative.
8.5.2 Non-Serious Adverse Event Reporting Requirements
Non-serious adverse events are to be reported on the adverse event CRFs, which are to be submitted to VIVUS, Inc. or its designee.
8.5.3 ***
If any trial subject becomes or is found to be *** while receiving the investigational product, the investigator must submit this information to VIVUS, Inc. on an ***.
The investigator will follow the *** and then notify VIVUS, Inc. or its designee of the outcome. The investigator will provide this information as a follow up to the ***.
For reported ***. The status of an ***.
If *** meet the criteria for immediate classification as a serious adverse event ***, the investigator should follow the procedures for reporting serious adverse events. Similarly, any *** that are considered to be adverse events should be reported as such on the appropriate CRF. However, *** need not be reported as an adverse event if there is no associated adverse outcome.
For reporting purposes, *** should be reported as serious adverse events, but because the ***.
9 DATA ANALYSIS/STATISTICAL METHODS
9.1 Sample Size Determination
In a previous *** study evaluating the effects of VI-0521, subjects treated with a *** had a mean (SD) weight loss of *** for subjects receiving *** treated subjects. If similar standard deviations are achieved in the present trial, the planned sample size of at least 250 subjects per treatment group should provide *** power to detect these differences.
9.2 Efficacy Analysis
9.2.1 Analysis of Primary Endpoint
The primary hypotheses are:
***
The primary calculated endpoints for the trial are the percent weight loss at week 56 calculated as *** and the percentage of subjects achieving at least 5% weight loss at week 56.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
The intent to treat (ITT) population (Section 9.4) is the primary analysis population. For subjects who discontinue treatment prior to trial completion, every attempt will be made to have them return to the clinic at week 56 for a final weight assessment, regardless of when they discontinued treatment. For subjects who ***.
Comparisons between treatments of percent weight loss will be assessed using a *** with factors of *** and ***and with ***. Comparisons between treatments of the percentage of subjects with at least 5% weight loss will be assessed by ***, with *** and *** and ***. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant for each of the co-primary end points at ***, then the testing will proceed to the *** also at ***. If the statistical comparison is not significant at the *** for the *** when compared with ***, then the statistical test will be stopped and the *** will not be tested.
If both dose groups are significantly better than ***, then the *** and *** dose groups will be compared. The *** for the difference in the mean percent body weight reduction between treatment groups will be derived.
The cumulative probability distribution as a function of percentage change in body weight for each treatment group will be plotted.
9.2.2 Analysis of Secondary Endpoints
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization and end of treatment (week 56) for *** and *** groups.
· The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups.
· The difference in change in waist circumference (randomization to week 56) for *** and *** groups.
The ITT population will be employed to evaluate all secondary efficacy endpoints, and a step down strategy analogous to that used for the primary endpoint will be implemented to protect the overall alpha levels for these analyses.
The difference in absolute weight reduction and the difference in change in waist circumference at week 56 from baseline will also be compared using the same *** as the primary end point. ***, with ***and *** and ***, will be used to compare the probability of reaching 10% body weight reduction from randomization (baseline) to week 56 between treatment groups.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
9.3 Analysis of Other Endpoints
9.3.1 Other Efficacy Endpoints
The changes in primary and secondary outcomes over monthly intervals during the study will be evaluated. The methodology for these comparisons will be similar to that used for primary and secondary endpoints. Details of these analyses will be provided in the Statistical Analysis Plan.
The *** data will consist of ***, each of which will be analyzed separately. Changes from baseline will be summarized using *** and ***. Effect sizes (change divided by baseline standard deviation) will also be summarized. Changes from baseline will be compared between treatment groups using *** as appropriate for *** assessments of ***. Results of the *** questions will be calculated for each study group and compared.
Changes in BMI between baseline and Visits 10 and 17 (weeks 28 and 56) and change in Framingham 10-year risk assessments between baseline and Visits 10 and 17 (weeks 28 and 56) will be evaluated. For these analyses, BMI and Framingham risk scores will be calculated by the central analysis group.
The change from baseline to Visit 17 (week 56) in the number and dosage of medications used to treat cardiovascular or metabolic risk factors will be calculated.
In the *** of subjects treated at ***, changes from *** to *** and *** in percent lean body mass and percent *** will be evaluated. Differences between treatment groups will be evaluated using methods similar to those used to evaluate other continuous variables. For these evaluations, data will be pooled from protocols OB-302 and OB-303.
9.3.2 *** Analysis
*** will be obtained during this trial using a multiple trough sampling scheme, where samples are obtained from each subject at ***. These data will be combined for analyses with data from other Phase 3 trials that will utilize similar sampling schemes. *** analyses will characterize the *** for both drugs. The effects of various covariates including (but not limited to) ***, gender, race, *** and age will be evaluated.
9.4 Analysis Populations
Intent-to-treat (ITT) and safety populations: All subjects who are randomized, take one or more doses of study medication and have at least one post dosing assessment will be included in the ITT (efficacy assessment) and safety (safety assessment) populations.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
9.5 ***
For those subjects who ***. If a subject ***.
9.6 ***
***
9.7 Safety Analysis
Safety analyses will be performed on the safety population and will include all data on all doses reported during the study.
9.7.1 Adverse Events
Adverse events will be coded using a MedDRA coding dictionary. The number and percentage of subjects who reported at least one adverse event in each system organ class and preferred term category, and the total number and percentage of subjects with any AE over all system organ classes will be summarized by treatment group.
Subsets of AEs that are considered serious or required discontinuation of the study medication will be listed by subject and presented separately.
9.7.2 Clinical Laboratory Tests
A summary of observed values and change from baseline will be presented for all laboratory parameters with numerical measures. For selected laboratory parameters, scatter plots of baseline versus week 56 results, will be produced by treatment group.
A laboratory value that is above or below normal range will be considered an abnormal value. For selected laboratory parameters, threshold limits of clinical concern will be defined as multiplicative factors of the normal ranges. The list of multiplicative factors for each laboratory parameter will be included in the Statistical Analysis Plan. The frequency and percentage of subjects with laboratory results above or below the normal range and threshold limits at each scheduled assessment or any time during the treatment will be summarized by treatment group.
9.7.3 ▇▇▇▇▇ ▇▇▇▇▇ and Other Safety Evaluations
Mean blood pressures, heart rate, respiration rate and temperature, obtained at each visit, will be summarized and plotted by treatment group. Medications, other than study medication, taken during the study will be considered as concomitant medications. Variables will be summarized by treatment group according to preferred terms using the World Health Organization (WHO) Drug Dictionary.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
9.7.4 Questionnaire Assessments
Changes in total *** will be summarized by treatment group *** and ***. Since the proposed indication for the study medication is weight loss, ***. Descriptive summaries of individual questionnaire items will also be presented.
9.8 Interim Analysis
An interim analysis may be conducted once approximately half of the study subjects have completed the intended course of treatment to provide information for the planning of future clinical trials that may be designed or commenced prior to the completion of this trial. No changes will be made to the conduct of the present study, including premature termination or increased sample size, based on the results of this interim analysis and no statistical adjustments for this interim analysis, if conducted.
9.9 ***
***
10 Quality control and quality assurance
During trial conduct, VIVUS, Inc. or its agent will conduct periodic monitoring visits to ensure that the protocol and good clinical practices (GCP) are being followed. The monitors may review source documents to confirm that the data recorded on CRFs are accurate. The investigator and institution will allow VIVUS, Inc. monitors or its agents and appropriate regulatory authorities direct access to all appropriate source documents to perform this verification.
The trial site and trial-related documents may be subject to review by the institutional review board (IRB)/independent ethics committee (IEC), and/or to quality assurance audits performed by VIVUS, Inc. or its agents and/or to inspection by appropriate regulatory authorities. Refer to Section 13.
It is important that the investigator(s) and their relevant personnel are available during monitoring visits and possible audits or inspections and that sufficient time is devoted to the process by the investigator and site personnel.
11 DATA HANDLING AND RECORD KEEPING
11.1 Case Report Forms / Electronic Data Record
As used in this protocol, the term case report form (CRF) should be understood to refer to either a paper form or an electronic data record or both, depending on the data collection method used in this trial.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
A CRF is required and should be completed for each included subject. The completed original CRFs are the sole property of VIVUS, Inc. and should not be made available in any form to third parties, except for authorized representatives of VIVUS, Inc. or appropriate regulatory authorities, without written permission from VIVUS, Inc.
It is the investigator’s responsibility to ensure CRF completion and to review and approve all CRFs. CRFs must be signed by the investigator or by an authorized staff member. These signatures serve to attest that the information contained on the CRFs is complete and accurate. At all times, the investigator has final personal responsibility for the accuracy and authenticity of all clinical and laboratory data entered on the CRFs. Subject source documents are the physician’s subject records maintained at the trial site. In most cases, the source documents will be the hospital’s or the physician’s chart. In cases where the source documents are the hospital or the physician’s chart, the information collected on the CRFs must match those charts.
11.2 Record Retention
To enable evaluations and/or audits from regulatory authorities or VIVUS, Inc. the investigator will keep records, including the identity of all participating subjects (sufficient information to link records, e.g., CRFs and hospital records), all original signed informed consent forms, copies of all CRFs, serious adverse event forms, source documents, and detailed records of treatment disposition. The records should be retained by the investigator according to specifications in the ICH guidelines, local regulations, or as specified in the Clinical Study Agreement, whichever is longer. The investigator must obtain written permission from VIVUS, Inc. before disposing of any records, even if retention requirements have been met.
If the investigator relocates, retires, or for any reason withdraws from the trial, VIVUS, Inc. should be prospectively notified. The trial records must be transferred to an acceptable designee, such as another investigator, another institution, or to VIVUS, Inc.
12 ETHICS
12.1 Institutional Review Board (IRB)/Independent Ethics Committee (IEC)
Regulations require that an IRB/IEC oversee all investigational drug studies. This board or committee, the makeup of which must conform to local, regional and national regulations, will approve all aspects of the study, including the protocol, advertising and written informed consent form to be used prior to initiation of the study. It is the responsibility of the investigator to have prospective approval of the trial protocol, protocol amendments, informed consent forms and other relevant documents, e.g., advertisements, if applicable, from the IRB/IEC. All correspondence with the IRB/IEC should be retained in the Investigator File. Copies of IRB/IEC approvals should be forwarded to VIVUS, Inc. or designee. All correspondence with the IRB/IEC should be retained in the Investigator file and copies forwarded to VIVUS, Inc. or designee.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
All amendments to the protocol must be reviewed and approved by VIVUS, Inc. and the IRB/IEC prior to implementation. The only circumstance in which an amendment may be initiated prior to IRB/IEC approval is where the change is necessary to eliminate apparent immediate hazards to the subjects. In that event, the investigator must notify the IRB/IEC and VIVUS, Inc. in writing within 5 working days after the implementation.
The investigator is responsible for obtaining annual (at a minimum) IRB/IEC renewal for the duration of the study. The investigator is also responsible for keeping the IRB/IEC advised of the progress of the study, of any changes made to the protocol as deemed appropriate, but at least once a year. Copies of the investigator’s report and of the IRB/IEC extension approval must be forwarded to VIVUS, Inc. or designee.
12.2 Ethical Conduct of the Trial
The trial will be performed in accordance with the protocol, International Conference on Harmonization Good Clinical Practice (ICH-GCP) guidelines and applicable local regulatory requirements and laws.
12.3 Subject Information and Consent
The informed consent form and any changes to the informed consent form made during the course of the trial must be agreed to by VIVUS, Inc. or designee and the IRB/IEC prior to its use and must be in compliance with all ICH-GCP, local regulatory requirements and legal requirements.
The investigator must ensure that each trial subject is fully informed about the nature and objectives of the trial and possible risks associated with participation and must ensure that the subject has been informed of his/her rights to privacy. The investigator will obtain written informed consent from each subject before any trial-specific activity is performed and should document in the source documentation that consent was obtained prior to enrollment in the trial. The original signed copy of the informed consent form must be maintained by the investigator and is subject to inspection by a representative of VIVUS, Inc., their representatives, auditors, the IRB/IEC and/or regulatory agencies. A copy of the signed informed consent form will be given to the subject.
12.4 Disclosure of Data
Data generated by this trial must be available for inspection by the U.S. Food and Drug Administration (FDA), by the sponsor or a designate acting on behalf of the sponsor, by applicable foreign health authorities, and by the IRB or IEC as appropriate. At a subject’s request, medical information may be given to their personal physician or other appropriate medical personnel responsible for their welfare.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Subject medical information obtained during the course of this trial is confidential and disclosure to third parties other than those noted above is prohibited.
13 REGULATORY CORRESPONDENCE
The trial site and trial-related documents may be subject to review by the IRB/IEC and/or to quality assurance audits performed by VIVUS, Inc. or its designee and/or to inspection by the FDA and/or applicable foreign health authorities. The investigator will notify VIVUS, Inc. or designee within *** working days following any FDA or other regulatory agency contact with the investigative site regarding this study. The investigator will provide VIVUS, Inc. with copies of all correspondence with the FDA or other regulatory agency which may affect the review of the current study (e.g., Form 483, Inspection Observations) or their qualification as an investigator in studies conducted by VIVUS, Inc. (e.g., warning letters).
14 DEFINITION OF END OF TRIAL
The end of trial is defined as the date when the last subject completes the last trial visit.
Additionally, data and materials that are required by the Sponsor before any trial site’s activity can be considered complete include:
· All completed Case Report Forms, appropriately signed by the investigator;
· All laboratory findings, clinical data and special test results collected during the trial period;
· Completed drug accountability and investigational materials return records;
· Statement of outcome for any serious adverse events reported during the study;
· Copy of notification to IRB or IEC indicating study completion.
15 SPONSOR DISCONTINUATION CRITERIA
Premature termination of this clinical trial may occur because of a regulatory authority decision, change in opinion of the IRB/IEC, drug safety problems, or at the discretion of VIVUS, Inc. In addition, VIVUS, Inc. retains the right to discontinue development of VI-0521 at any time.
If a trial is prematurely terminated or discontinued, VIVUS, Inc. will promptly notify the investigator. After notification, the investigator must contact all participating subjects within 15 days. As directed by VIVUS, Inc. or its designee, all trial materials must be collected and all CRFs completed to the greatest extent possible.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
16 PUBLICATION OF TRIAL RESULTS
All information and data, including the terms of this protocol, and all data, clinical results, and research conducted hereunder concerning VIVUS, Inc’s products and operations including VIVUS, Inc. patent applications, formulas, manufacturing processes, basic scientific data, and formulation information that has been supplied by VIVUS, Inc. and not previously published are considered confidential by VIVUS, Inc. and will remain the sole property of VIVUS, Inc. The investigator understands and agrees that said proprietary and/or confidential information disclosed to or produced by him/her thereunder is highly valuable to VIVUS, Inc. and will be used exclusively by the investigator in accomplishing this study and will not be used for any other purposes without VIVUS. Inc’s prior written consent. The investigator agrees that he/she will not use any such proprietary and/or confidential information for any other purpose. The investigator also understands and agrees that such disclosure will not be deemed to grant to the investigator a license for use of said proprietary and/or confidential information, except as expressly provided herein.
It is understood by the investigator that the information developed in the clinical study will be used by VIVUS, Inc. in connection with the development of this product. This information, therefore, may be disclosed and used solely by VIVUS, Inc. as required to such third parties and agencies as VIVUS, Inc, in its sole discretion, warrants. In order to allow for the use of the information derived from the clinical studies, it is understood that there is an obligation to provide to VIVUS, Inc. complete test results and all data developed in this study. The investigator agrees to promptly answer all inquires from VIVUS, Inc. regarding completion, legibility or accuracy of trial data in the case report.
VIVUS, Inc. recognizes the value of disseminating research results and expects that publication of all results from this study will be undertaken by a collaborative group of study investigators who made significant contributions to the study design, the treatment of study subjects, and evaluation of study data. However, after 1) submission of the multicenter results for publication, 2) notification ***.
Investigators shall furnish the *** with a written copy of any proposed publication or other disclosure of study results (including disclosures at research seminars, lectures and professional meetings) *** prior to submission for publication or disclosure so that *** may have a reasonable opportunity to protect its proprietary rights to information, inventions, or products developed under this study and to insure that reported data are factually correct. Upon the ***, the investigator shall not publish or disclose information related to this study. Further, if the *** believes that such publication or disclosure contains confidential information, the investigator agrees to remove such confidential information from the proposed publication or disclosure.
VIVUS, Inc. agrees that before it publishes any results of this study in a refereed journal, it will provide the investigator, for review, a prepublication manuscript *** prior to the submission of the manuscript to the publisher.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
17 REFERENCES
(1) Adipex-P® (Phentermine Hydrochloride USP, 37.5 mg) Package Insert (2005). Gate Pharmaceuticals, Sellersville, PA.
(2) ▇▇▇▇▇▇ D, ▇▇▇ M. Obesity and Regulation of Body Mass. Lehninger Principles of Biochemistry. 4th ed. New York: ▇▇ ▇▇▇▇▇▇▇ and Company; 2005:912-913.
(3) Montague C, Farooqi I, ▇▇▇▇▇▇▇▇▇ J, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997;387:903-908.
(4) Heymsfield S, ▇▇▇▇▇▇▇▇▇ A, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: A randomized, controlled, dose-escalation trial. JAMA 1999;282:1568-1575.
(5) Topamax® (topiramate) Tablets and Topamax® (topiramate capsules) Sprinkle Capsules Package Insert (June 2003). Ortho-▇▇▇▇▇▇ Pharmaceutical, Inc., ▇▇▇▇▇▇▇, ▇▇ ▇▇▇▇▇.
(6) ▇▇▇▇▇▇ K, ▇▇▇▇▇▇▇ M, ▇▇▇▇▇ C, ▇▇▇▇▇▇▇▇ C. Prevalence and trends in obesity among US adults, 1999-2000. JAMA 2002;288:1723-1727.
(7) ▇▇▇▇▇▇▇ P, ▇▇▇▇▇ T, ▇▇▇▇ G, et al. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss: An update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Commission of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 2006;113:898-918.
(8) ▇▇▇▇▇ C, ▇▇▇▇▇▇▇ M, ▇▇▇▇▇▇ L, et al. Prevalence of overweight and obesity in the ▇▇▇▇▇▇ ▇▇▇▇▇▇, ▇▇▇▇-▇▇▇▇. JAMA 2006;295:1549-1555.
(9) Must A, Spadano J, ▇▇▇▇▇▇▇ E, et al. The disease burden associated with overweight and obesity. JAMA 1999:282;1523-1529.
(10) Katzmarzyk P, ▇▇▇▇▇▇▇ I, Ardern C. Physical inactivity, excess adiposity and premature mortality. Obes Res 2003;4:257-290.
(11) ▇▇▇▇▇ K, ▇▇▇▇▇▇▇▇▇ A, ▇▇▇▇▇ T, et al. Overweight, obesity, and mortality in a large prospective cohort of persons 50 to 71 years old. NEJM 2006;355:763-778.
(12) ▇▇▇▇▇▇▇▇▇ D. Beneficial health effects of modest weight loss. Int J Obes 1992;6:297-415.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
(13) Pasanisi F, Contaido F, ▇▇▇▇▇▇▇▇ G, ▇▇▇▇▇▇▇ M. Benefits of sustained moderate weight loss in obesity. Nutr Metab Cardiovasc Dis 2001;11:401-406.
(14) Douketis J, ▇▇▇▇▇ C, Thabane L, ▇▇▇▇▇▇▇▇▇▇ D. Systematic review of long-term weight loss studies in obese adults: Clinical significance and applicability to clinical practice. Int J Obes 2005;10:1153-1167.
(15) ▇▇▇▇▇▇ R, Majumdar S. Drug treatments for obesity: Orlistat, sibutramine, and rimonabant. Lancet 2007;369:71-77.
(16) ▇▇▇-▇▇▇▇▇▇▇▇ E, ▇▇▇▇▇▇▇ M, ▇▇▇▇▇▇▇▇ E, et al. Predictors of weight loss in adults with topiramate-treated epilepsy. Obes Res 2003;11:556-562.
(17) ▇▇▇▇ G, ▇▇▇▇▇▇▇▇▇ P, ▇▇▇▇▇ S, et al. A 6-month randomized, placebo-controlled, dose-ranging trial of topiramate for weight loss in obesity. Obes Res 2003;11:722-733.
(18) ▇▇▇▇▇▇▇ J, ▇▇▇▇▇▇▇ L, ▇▇▇▇▇▇▇▇ A, et al. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of topiramate in the treatment of obese subjects. Int J Obes Relat Metab Disord 2004;28:1399-1410.
(19) Clinical Study Report: A Phase II, 24-Week, Randomized, Double-Blind, Placebo-Controlled, Single-Center Study to Examine Safety, Tolerability and Efficacy of Topiramate and Phentermine Combination Therapy for Weight Loss in Healthy Obese Subjects. Data on file. VIVUS, Inc., Mountain View, CA.
(20) The LEARN® Program for Weight Management 2000. ▇▇▇▇▇ ▇. ▇▇▇▇▇▇▇▇. The Life Style Company™. Dallas.
(21) National Heart, Lung and Blood Institute Obesity Education Initiative. The Practical Guide: Identification, Evaluation and Treatment of Overweight and Obesity in Adults. NIH Publication No. 00-4084. October 2000.
(22) ***
(23) ***
(24) ***
(25) ***
(26) ***
(27) ***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
(28) ▇▇▇▇▇▇ P, ▇’▇▇▇▇▇▇▇▇ R, ▇▇▇▇ D, et al. Prediction of Coronary Heart Disease Using Risk Factor Categories. Circulation 1998;97:1837-1847.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 1: SCHEDULE OF STUDY ACTIVITIES
|
|
|
*** |
|
*** |
|
*** |
|
*** | ||||||||||||||||||||||||||
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
*** |
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 2: ***
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 3: ***
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 4: PROTOCOL AMENDMENTS
Amendment Tracking
|
Protocol Title: |
|
A Phase III Randomized, Double-Blind, Placebo-Controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in an Adult Population with BMI > *** |
|
|
|
|
|
Protocol Number: |
|
OB-302 |
Indicate any amendments to this protocol by writing the Amendment Number and the Date of the Amendment in the space below. This page will be used to track the original protocol and its amendments. This is the original protocol if no amendment number is listed below.
Amendment Number 3
Date of Amendment: ***
(19 items)
Rationale: This protocol amendment is being implemented at the request of the FDA to incorporate assessments of *** and *** using the ***, and assessments of *** using the *** in all study subjects at ***. Previously, follow-up *** evaluations were done only if other *** assessments revealed a ***. This amendment also provides for body composition assessments at selected sites using ***. These changes are not anticipated to have any impact on the safety of study subjects.
|
Section and/or |
|
Protocol |
|
Amendment 3, *** |
|
Protocol Synopsis: Efficacy Endpoints - Additional efficacy endpoints include: |
|
*** |
|
Added: “Effect on body composition, as evaluated by ***. *** assessments will be performed at only at a ***.” |
|
|
|
|
|
|
|
Protocol Synopsis: Safety Endpoints |
|
*** |
|
Change: “Follow-up *** assessments will be done at ***, and a single question assessment of *** and *** will be asked at ***.”
To: “Follow-up assessments will be done at *** after ***.” |
|
|
|
|
|
|
|
List of Abbreviations |
|
*** |
|
Added: “***” |
|
|
|
|
|
|
|
Rationale |
|
*** |
|
Change: “VI-0521 is an exploratory weight loss therapy that is a new combination of two currently approved drugs, phentermine and topiramate.”
To: “VI-0521 is an investigational weight loss therapy that is a combination of two currently approved drugs, phentermine and topiramate.” |
|
|
|
|
|
|
|
Trial Design: Secondary |
|
*** |
|
Change: “Change in BMI between baseline and week 28 and end of treatment |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
efficacy endpoints are: |
|
|
|
(Visit 17, week 56) will be evaluated. .”
To: “Change in BMI between baseline and week 28 and end of treatment (Visit 17, week 56) will be evaluated. Additionally, body composition will be assessed by *** at ***. These evaluations will be made at ***, and at *** and ***.”
Change: “Safety evaluations will include assessment of adverse events, including eye symptoms, ***.”
To: “Safety evaluations will include assessment of adverse events, including eye symptoms, ***.” |
|
|
|
|
|
|
|
Trial Period: Randomization (Visit 2) |
|
*** |
|
Added: “Obtain written informed consent for *** procedures and perform *** scan ***. *** scans may be performed between Visits 1 and 2, after results from Visit 1 indicate subject may be eligible to participate;”
Change: “Assess adverse events (including eye symptoms and ***), if any;”
To: “Assess adverse events (including eye symptoms), if any;” |
|
|
|
|
|
|
|
Trial Period: Titration (Visit 3) |
|
*** |
|
Added: “Administer *** and ***;”
Change: “Assess adverse events (including eye symptoms and ***), if any;”
To: “Assess adverse events (including eye symptoms), if any;” |
|
|
|
|
|
|
|
Trial Period: Treatment (Visits 4 through 16) |
|
*** |
|
Added: “Administer *** and ***;”
Delete: “Administer *** (Visits 7, 10, and 13 only);”
Change: “Assess adverse events (including eye symptoms and ***), if any;”
To: “Assess adverse events (including eye symptoms), if any;”
Added: “Perform *** scan *** (Visit 10 only);” |
|
|
|
|
|
|
|
End of Treatment (Visit 17 or Withdrawal from Study) |
|
*** |
|
Added: “Administer ***;”
Change: “Assess adverse events (including eye symptoms and ***), if any;”
To: “Assess adverse events (including eye symptoms), if any;”
Added: “Perform *** scan ***;” |
|
|
|
|
|
|
|
Subject Withdrawal |
|
*** |
|
Change: “At about the 56 week time point, withdrawn subjects who have not continued site visits should be asked to return to the site to obtain weight and waist circumference measurements at a minimum and, if possible, the following additional information: adverse events, ▇▇▇▇▇ ▇▇▇▇▇, laboratory tests (chemistry, hematology, urinalysis), concomitant medications, questionnaires ***.”
To: “At about the 56 week time point, withdrawn subjects who have not continued site visits should be asked to return to the site to obtain weight and |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
waist circumference measurements at a minimum and, if possible, the following additional information: adverse events, ▇▇▇▇▇ ▇▇▇▇▇, laboratory tests (chemistry, hematology, urinalysis), concomitant medications, questionnaires ***.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “The *** is being used ***. This questionnaire will be completed at *** and at ***.”
To: “The *** is being used ***. This questionnaire will be completed at ***, and at *** after the ***.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “Subsequent *** evaluations will be done only in *** who demonstrate a ***.”
To: “Subsequent *** evaluations will be done at *** after ***. All *** assessments must be administered by a trained interviewer. If any assessments reveal ***, then the results must be reviewed by a physician investigator prior to ***.” |
|
|
|
|
|
|
|
Body Composition |
|
*** |
|
Added new section:
“At a selected subset of study sites, body composition assessments will be made at baseline (Visit 2), week 28 (Visit 10) and week 56 (Visit 17) using ***. Scans will be collected at a sufficient number of study sites to provide body composition data on approximately *** subjects from this study combined with protocol OB-303. Equipment and procedures used to obtain *** data will be standardized as described in a separate document. All sites will be trained on these procedures prior to performing scans on study subjects. Scans will be read at a central facility and the reader will be blinded to the identity of the subjects, the treatment assignment, and the visit number.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “All subjects will be screened for the presence and *** at *** and subsequently at regular intervals throughout the study: ***, and *** using a validated survey instrument *** designed for assessment of *** in a primary care setting.”
To: “All subjects will be screened for the presence and *** at *** and subsequently at *** after the *** using a validated survey instrument *** designed for assessment of *** in a primary care setting.”
Change: “The *** is a *** module based directly on the diagnostic criteria for ***. At study visits where *** assessments are not included, subjects will be asked the following question to assess ***.”
To: “The *** is a *** module based directly on the diagnostic criteria for ***. *** will also be assessed at *** and at *** following the *** using the ***.”
Change: “Any ***. Investigators are encouraged to administer the *** on an ad-hoc basis as part of the clinical assessment of ***. These ad-hoc questionnaires will be maintained as source documentation, but will not be analyzed as a separate ***.”
To: “Any *** must be ***.” |
|
|
|
|
|
|
|
Other Efficacy Endpoints |
|
*** |
|
Added: “In the *** of subjects treated at ***, changes from *** to *** and *** in percent lean body mass and percent *** will be evaluated. |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
Differences between treatment groups will be evaluated using methods similar to those used to evaluate other continuous variables. For these evaluations, data will be pooled from protocols OB-302 and OB-303.” |
|
|
|
|
|
|
|
Appendix 1: Schedule of Study Activities |
|
*** |
|
Added: “***” and only marked at *** |
|
|
|
|
|
|
|
Appendix 1: Schedule of Study Activities — *** |
|
*** |
|
Added at *** |
|
|
|
|
|
|
|
Appendix 1: Schedule of Study Activities — *** |
|
*** |
|
Added at *** |
|
|
|
|
|
|
|
Appendix 1: Schedule of Study Activities — *** |
|
*** |
|
Change: “***”
To: “***” |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
Protocol Title: |
|
A Phase III Randomized, Double-Blind, Placebo-Controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in an Adult Population with BMI > *** |
|
|
|
|
|
Protocol Number: |
|
OB-302 |
Indicate any amendments to this protocol by writing the Amendment Number and the Date of the Amendment in the space below. This page will be used to track the original protocol and its amendments. This is the original protocol if no amendment number is listed below.
Amendment Number 2
Date of Amendment: ***
(10 items)
Rationale: This amendment is being implemented to address inconsistencies in the description of various study exclusions and analyses, and to specify that an ***. Various typographical errors in the previous version of the protocol have also been corrected. None of the changes implemented with this amendment are anticipated to have any impact on safety risks to the study subjects.
|
Section and/or |
|
Protocol |
|
Amendment 2, *** |
|
Protocol Synopsis: Study Subjects |
|
*** |
|
Change: “Major exclusion criteria include: type 2 diabetes; known or suspected valvular heart disease;”
To: “Major exclusion criteria include: type 2 diabetes; known or suspected clinically significant valvular heart disease;” |
|
|
|
|
|
|
|
Protocol Synopsis: Statistical Methods |
|
*** |
|
Change: “Comparisons between treatments will be assessed using a *** with factors of *** and ***and with ***. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant at ***, then the test will proceed to the *** also at the ***.”
To: “Comparisons between treatments will be assessed using a *** with factors of *** and *** and with *** for percent weight loss, and by *** for percent of subjects achieving at least 5% weight loss. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant at *** for both co-primary endpoints, then the test will proceed to the *** also at the ***.” |
|
|
|
|
|
|
|
Background |
|
*** |
|
Change: “Obesity is associated with numerous co-morbidities including dyslipidemia, coronary artery disease, hypertension, stroke and type 2 diabetes.(7), (9) Epidemiological data indicate that obesity is associated with increased mortality,(10) and a recent study of over 500,000 individuals concluded that excess body weight during midlife, including overweight, was associated with an increased risk of death.(11)”
To: “Obesity is associated with numerous co-morbidities including dyslipidemia, |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
coronary artery disease, hypertension, stroke and type 2 diabetes.(7), (9) Epidemiological data indicate that obesity is associated with increased mortality,(10) and a recent study of over 500,000 individuals concluded that excess body weight during midlife was associated with an increased risk of death.(11) “ |
|
|
|
|
|
|
|
Trial Design |
|
*** |
|
Change: “In this prospective, randomized, double blind, placebo-controlled prospective trial, subjects meeting the eligibility criteria will be randomly assigned (with equal probability) among the *** treatment groups described in Figure 1.”
To: “In this prospective, randomized, double blind, placebo-controlled trial, subjects meeting the eligibility criteria will be randomly assigned in a ratio of *** among the *** treatment groups described in Figure 1.” |
|
|
|
|
|
|
|
Exclusion Criteria #15 |
|
*** |
|
Change: “Unstable angina, congestive heart failure (NYHA Class II, III or IV), or suspected or known cardiac valvulopathy;”
To: “Unstable angina, congestive heart failure (NYHA Class II, III or IV), or known or suspected clinically significant cardiac valvulopathy;” |
|
|
|
|
|
|
|
Titration: (Visit 3) |
|
*** |
|
Change: “Review the study medication used during weeks 1 and 2; assess treatment compliance and perform drug accountability. Return the card to the subject for use during weeks 3 and 4;”
To: “Review the study medication used during weeks 1 and 2; assess treatment compliance and perform drug accountability. Return the titration card to the subject for use during weeks 3 and 4;” |
|
|
|
|
|
|
|
Analysis of Primary Endpoint |
|
*** |
|
Change: “The primary calculated endpoints for the trial are based on the percent weight loss at week 56 calculated as *** and the percentage of subjects achieving at least 5% weight loss at week 56.
The intent to treat (ITT) population (Section 9.4) is the primary subject population. For subjects who discontinue treatment prior to trial completion, every attempt will be made to have them return to the clinic at week 56 for a final weight assessment, regardless of when they discontinued treatment. For subjects who ***.
Comparisons between treatments will be assessed using a *** with factors of *** and *** and with ***. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant for the primary end points at ***, then the test will proceed to the *** also at ***.”
To: “The primary calculated endpoints for the trial are the percent weight loss at week 56 calculated as *** and the percentage of subjects achieving at least 5% weight loss at week 56.
The intent to treat (ITT) population (Section 9.4) is the primary analysis population. For subjects who discontinue treatment prior to trial completion, every attempt will be made to have them return to the clinic at week 56 for a final weight assessment, regardless of when they discontinued treatment. For subjects who ***.
Comparisons between treatments of percent weight loss will be assessed using a *** with factors of *** and *** and with ***. Comparisons between treatments of the percentage of subjects with at least 5% weight loss will be assessed by ***, with *** and *** and ***. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant for |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
each of the co-primary end points at ***, then the testing will proceed to the *** also at ***.” |
|
|
|
|
|
|
|
Analysis of Secondary Endpoint |
|
*** |
|
Change: “The difference in absolute weight reduction and the difference in change in waist circumference at week 56 from baseline will also be compared using the same *** as the primary end point. ***, with *** and *** and ***, will be used to compare the probability to reach 5% and 10% body weight reduction between randomization (baseline) and week 56 between treatment groups.”
To: “The difference in absolute weight reduction and the difference in change in waist circumference at week 56 from baseline will also be compared using the same *** as the primary end point. ***, with *** and *** and ***, will be used to compare the probability of reaching 10% body weight reduction from randomization (baseline) to week 56 between treatment groups.” |
|
|
|
|
|
|
|
Adverse Events |
|
*** |
|
Change: “Adverse events will be coded using a MedDRA coding dictionary. The number and percentage of subjects who reported at least one adverse event in each organ class and preferred term category, and the total number and percentage of subjects with any AE over all organ system will be summarized by treatment group.”
To: “Adverse events will be coded using a MedDRA coding dictionary. The number and percentage of subjects who reported at least one adverse event in each system organ class and preferred term category, and the total number and percentage of subjects with any AE overall system organ classes will be summarized by treatment group.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “No *** will be employed for this study.”
To: “***” |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
Protocol Title: |
|
A Phase III Randomized, Double-Blind, Placebo-Controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in an Adult Population with BMI > *** |
|
|
|
|
|
Protocol Number: |
|
OB-302 |
Indicate any amendments to this protocol by writing the Amendment Number and the Date of the Amendment in the space below. This page will be used to track the original protocol and its amendments. This is the original protocol if no amendment number is listed below.
Amendment Number 1
Date of Amendment: ***
(14 items)
This protocol amendment is being implemented to add a co-primary endpoint to the study analysis section and to add additional laboratory tests to the schedule of evaluations. The definition of child-bearing potential has also been clarified, and miscellaneous typographical errors have been corrected. These changes are being made at the request of the FDA, and are not anticipated to significantly affect the risk to study subjects.
|
Section and/or |
|
Protocol |
|
Amendment 1, *** |
|
Protocol Synopsis: Efficacy Endpoints |
|
*** |
|
Change: “The primary endpoint is the differences between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), and percent of subjects with weight loss of 5% or more at end of treatment (week 56). Percent weight loss will be calculated as ***.
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) between *** and *** groups;
· The difference in percent of subjects who achieve a reduction in total body weight of at least 5% and at least 10% between randomization (baseline) and end of treatment (week 56) between *** and *** groups; and”
To: “The primary endpoints are the differences between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), and percent of subjects with weight loss of 5% or more at end of treatment (week 56). Percent weight loss will be calculated as ***.
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) between *** and *** groups; |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
· The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) between *** and *** groups; and” |
|
|
|
|
|
|
|
Trial Design |
|
*** |
|
Change: “The primary endpoint is the difference between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), Percent weight loss will be calculated as ***. Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) between *** and *** groups;
· The difference in percent of subjects who achieve a reduction in total body weight of at least 5% and at least 10% between randomization (baseline) and end of treatment (week 56) between *** and *** groups; and”
To: “The primary endpoints are the differences between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), and percent of subjects with weight loss of 5% or more at end of treatment (week 56). Percent weight loss will be calculated as ***. Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) between *** and *** groups;
· The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) between *** and *** groups; and” |
|
|
|
|
|
|
|
Inclusion Criteria |
|
*** |
|
Change: “Females are considered to be of child-bearing potential unless they have undergone a hysterectomy or bilateral oophorectomy, have a documented follicle stimulating hormone level >40 IU/L or are 55 years of age or greater and have experienced cessation of menses for at least 1 year.”
To: “Females are considered to be of child-bearing potential unless they have undergone a hysterectomy or bilateral oophorectomy, are 55 years of age or greater and have experienced spontaneous cessation of menses for at least 1 year, or have a documented follicle stimulating hormone level >40 IU/L.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “When *** is not appropriate or when *** may be required due to events unrelated to subject treatment, subjects may ***. If dosing has been ***, a new titration kit should be ordered through *** to ***.”
To: “When *** is not appropriate or when *** may be required due to events unrelated to subject treatment, subjects may ***. *** are possible with agreement from the medical monitor. All subjects undergoing *** for *** may be *** based on discretion of the PI. If dosing has been ***, a new titration kit should be ordered through ***to ***.” |
|
|
|
|
|
|
|
Randomization (Visit 2) |
|
*** |
|
Add: “Obtain blood sample for laboratory testing (biomarkers);”
Change: “Assess adverse events (including eye symptoms), if any;
To: “Assess adverse events (including eye symptoms and ***), if any;” |
|
|
|
|
|
|
|
Titration (Visit 3) |
|
*** |
|
Change: “Assess adverse events (including eye symptoms), if any; |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
To: “Assess adverse events (including eye symptoms and ***), if any;” |
|
|
|
|
|
|
|
Treatment (Visit 4-16) |
|
*** |
|
Change: “Assess adverse events (including eye symptoms), if any;
To: “Assess adverse events (including eye symptoms and ***), if any;” |
|
|
|
|
|
|
|
End of Treatment (Visit 17 or withdrawal from study) |
|
*** |
|
Change: “Assess adverse events (including eye symptoms), if any;
To: “Assess adverse events (including eye symptoms and ***), if any;” |
|
|
|
|
|
|
|
Biomarkers |
|
*** |
|
Added new section: “Blood biomarkers including C-reactive protein, adiponectin, and fibrinogen will be evaluated at basline (Visit 2) and end of treatment (Visit 17 or study withdrawal). All post-screening biomarker results will be blinded to investigators and sponsor.” |
|
|
|
|
|
|
|
Analysis Primary Endpoint |
|
*** |
|
Change: “***
The primary calculated endpoints for the trial are based on the percent weight loss at week 56 calculated as ***.”
To: “***
The primary calculated endpoints for the trial are based on the percent weight loss at week 56 calculated as *** and the percentage of subjects achieving at least 5% weight loss at week 56. “ |
|
|
|
|
|
|
|
Analysis of Secondary Endpoint |
|
*** |
|
Change: “The difference in percent of subjects who achieve a reduction in total body weight of at least 5% and at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups. “
To: “ The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups. “ |
|
|
|
|
|
|
|
Interim Analysis |
|
*** |
|
Change: “No changes will be made to the conduct of the present study, including premature termination or increased sample size, based on the results of this interim analysis and no statistical adjustments for this interim analysis, if conducted, will be made.
To: “No changes will be made to the conduct of the present study, including premature termination or increased sample size, based on the results of this interim analysis and no statistical adjustments for this interim analysis, if conducted.” |
|
|
|
|
|
|
|
Appendix 1 |
|
*** |
|
Added: “***” |
|
|
|
|
|
|
|
Appendix 1 |
|
*** |
|
Change: “***”
To: “***” |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #02
Appendix 2 – Scope of Work
VIVUS, Inc.
Qnexa OB-302
PROJECT OVERVIEW
▇▇-▇▇▇
▇▇▇ ▇▇-▇▇▇ trial is a phase III randomized, double blind, placebo controlled multicenter study to determine the safety and efficacy of VI-0521 in the treatment of obesity in otherwise healthy adults.
PROJECT TEAM
Study Management
The overall management of the study will be the responsibility of the Senior Clinical Trial Manager (CTM). The Senior CTM will oversee and coordinate the management of the study as well as oversee the study specific CTM. This oversight will ensure consistency and allow VIVUS Study Management to have one primary contact for the Qnexa program. The Medpace CTM assigned to OB-302 will work closely with the VIVUS Study Manager, Medpace Medical Expert, and VIVUS Clinical Leader to address protocol questions and interpretations while maintaining close oversight of study-related processes and documents. The OB-302 CTM will supervise all Clinical Research Associates (CRAs) and Project Coordinators assigned to the project.
The Project Coordinators will be responsible for day-to-day study management functions, including the generation of status reports, organization of supplies, generation and compilation of newsletters, and input of all study information into the ClinTrak® Study Management System, a web-based, proprietary research management system designed by Medpace. The Project Coordinators will organize teleconferences and team meetings, including the compilation of agendas and meeting minutes.
The Study Start-Up Manager and Study Start-Up Coordinators will work closely with the CTM and Project Coordinators to ensure sites become active in the most time effective manner.
The Medpace Contracts Attorney will be responsible for the execution of Investigator contracts (upon VIVUS defined process). The Contracts Attorney will work closely with the Start-Up Manager and Medpace CTM to ensure contracts are executed in a timely manner.
The Medpace Medical Expert assigned to this project will work closely with the VIVUS Clinical Leader. The Medpace Medical Expert will assist with protocol design and medical interpretation of entry criteria and adverse events (AEs). The Medical Expert will also be involved in the training of CRAs and other staff members participating in the project. The Medical Expert will review and approve the coding of concomitant medications, medical histories, AEs, and will provide the medical context for the statistical analysis and medical writing.
|
CONFIDENTIAL |
September 11, 2007 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
The Medical Expert will assist in the review of the protocol, train Medpace personnel internally as to the background of the study compound and design of the study, participate in the project teleconferences and meetings, work hand-in-hand with the OB-302 CTM, and have heavy involvement in the clinical study report. The Medical Expert’s role and decision making rights are dictated by VIVUS (e.g. inclusion/exclusion of patients, discussions with Investigators about withdrawing a patient, etc.). This decision making power often times reduces the oversight needed by the sponsor. For questions the OB-302 CTM is not comfortable answering, she will contact the Medical Expert for guidance. Obviously, VIVUS will be involved in study oversight based on pre-defined terms with the VIVUS Clinical Development Team. The Medpace Medical Expert is available 24 hours a day, 7 days a week via the Medpace Project Helpline.
Clinical Monitoring
Medpace operates in North America with a primarily centralized monitoring team of over 140 CRAs to promote greater standardization, cohesiveness, support, and stability. Each of the Medpace CRAs assigned to this project have monitoring experience and strong clinical backgrounds.
Clinical Safety
The Clinical Safety will be managed by VIVUS or its designee. VIVUS Clinical Leader to be involved with casualty assignment for all Serious Adverse Events (SAEs).
Data Management
A Data Manager will serve as the primary contact for the Data Management team. Data Coordinators will be involved in the day-to-day operations and report issues to the Data Manager. Data Entry Specialists and Database Programmers will also be utilized.
Biometrics
Key members of our Biometrics team include Biostatisticians and Statistical Analysts. The Biostatistician assigned to this project will develop the analysis plan and coordinate biometrics activities. The Lead Statistical Analyst will work closely with the Biostatistician to ensure a clear understanding of the analysis plan and communicate any programming issues that may arise.
Medical Writing
The Medical Writing team works in collaboration with the Medical Experts to prepare research reports meeting International Conference on Harmonisation (ICH) and Sponsor guidelines. All Medpace Medical Writers have extensive experience in regulatory submission preparation. The Medical Writing team is actively involved throughout the conduct of the trial.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Team Organization Chart
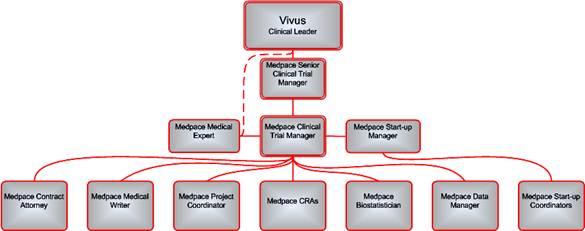
PROJECT START-UP
Protocol
VIVUS will prepare the protocol. Medpace will review the protocol and provide comments before it is finalized.
Case Report Forms
Medpace will design the electronic case report forms (eCRFs) for the trial, including completion instructions, according to the final protocols and the Medpace template. VIVUS must review and approve the eCRFs before they are finalized.
Project Initiation
Prior to the study site initiation visits, a project kickoff meeting will be held at Medpace involving Medpace and VIVUS personnel to review the study protocol, eCRFs, and overall project coordination. Medpace project team members and VIVUS personnel will participate in this meeting.
Interactive Voice Response System
Medpace will provide a customized (study-specific) interactive voice response system (IVRS) to provide patient randomization, and drug management. The Medpace IVRS is a proprietary in-house developed system. The system provides both voice and web access and has been developed in conjunction with our web based Clintrak® system providing seamless functionality throughout the conduct of the study. The VIVUS Team (no limit applied to number of team members) will have access to review reports within the IVRS. Medpace will perform the User Acceptance Testing (UAT) for each site.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
The IVRS will include:
· Subject Screens/Screen Failures;
· Subject randomization;
· Patient visit tracking;
· Inventory management (site supply set-up, initial bulk supply, and resupply of one additional shipment per patient);
· Notifications of site shipments;
· Confirmation of receipt of shipments; and
· Customized reports.
VIVUS will review IVRS and approve system prior to finalization.
Study Medication Supply and Storage
VIVUS will be responsible for the supply, packaging, labeling, storage, and destruction of study medication. Distribution of the study medication will be tracked and initiated via the Medpace IVRS. Study medication accountability procedures will follow Medpace standard operating procedures (SOPs) and utilize a study medication accountability log that has been approved by VIVUS.
Recruitment Oversight Plan
The Medpace CTM, in collaboration with VIVUS, will develop a recruitment oversight plan. Medpace understands the importance of rapid recruitment and the necessity to keep patients in the trial until completion. Medpace will develop processes prior to study initiation to ensure recruitment is efficient and retention of patients in the study is maximized. The plan will include details on:
· Initial collection of essential documents;
· Patient recruitment (including tools, site-specific plans, contingencies, etc.); and
· Patient recruitment tracking reports.
The tools noted above include:
· Inclusion/exclusion cards;
· Patient emergency cards;
· Enrollment tracking forms;
· Laminated patient visit schedule;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Pocket protocol; and
· Advertising tools (e.g., posters, table tents, language for newspaper/radio advertisements, etc.).
Medpace understands each site has a different experience and approach to patient recruitment. The Medpace CTM and Medpace CRAs will work with personnel from each site to help maximize their efforts. The project team maintains frequent contact during the active recruitment phase to assess each site’s activity and offer assistance when needed. Medpace acknowledges the importance of site recognition and offers various incentives throughout the recruitment period. Examples of site incentives are “Site of the Month” awards and weekly faxes displaying each site’s activity (often included in the newsletter as well). The time associated with these types of incentives are inclusive of the budget. However, often times, monetary incentives are built into the Investigator’s grant to encourage rapid essential document collection and/or timely recruitment.
Patient retention is vital to the success of a trial. The Medpace CTM and the Medpace CRAs will work with the sites to understand the needs and motivations for patients to remain in the study. They will help educate the sites on the importance of “customer service” and “patient satisfaction” as elements that ensure continued patient participation. Examples of patient retention methods include quarterly patient newsletters, ideas for site customer service, and tokens of appreciation for patients.
Site Selection and Pre-study Visits
VIVUS, in conjunction with Medpace, will identify qualified Investigators. VIVUS and Medpace will also work together to provide and negotiate Confidentiality Disclosure Agreements (CDAs) as well as create and evaluate site questionnaires. In not knowing the number of pre-study visits Medpace will be required to conduct, a unit price for a pre-study visit has been provided in the budget.
Pre-study visits will be conducted consistent with Medpace SOPs. Medpace will provide a pre-study visit report to VIVUS within 10 business days of each visit.
These visits will include, but are not limited to:
· Determining whether or not the site has clinical staff of appropriate education, training, and experience to manage the study and have sufficient capacity to perform the required tasks;
· Determining whether or not the site has appropriate facilities to conduct the study;
· Determining there are no competing studies that will conflict with patient enrollment and that the site has sufficient patients and processes to enroll patients in the time identified for patient recruitment; and
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Determining whether or not the site has appropriate resources and procedures to maintain appropriate records according to FDA requirements.
Study Start-up Team
The efficient start-up of the *** study sites for OB-302 will have a significant effect on patient recruitment time. Medpace will utilize its Study Start-up team for additional support during the initial phase of the trials to expedite the overall study start-up for each site.
The Study Start-up team works directly with the CTM and the Project Coordinators. The team is comprised of a Study Start-up Manager and several experienced Study Start-up Coordinators. The team is responsible for many of the key start-up activities, including:
· Submission to the central IRB;
· Coordination and tracking of essential documents packages for each site;
· Investigator meeting presentations and binders; and
· Site tools.
Central Laboratory Selection
Medpace Reference Laboratories (MRL) will be utilized for processing the clinical laboratory samples. MRL is committed to providing comprehensive laboratory services of the highest quality to the pharmaceutical and biotechnology industries.
Investigators’ Meeting
An Investigators’ Meeting will be held for the OB-302 study. VIVUS will arrange the meeting (including contracting with a third-party vendor) and Medpace will prepare the meeting materials, including preparation and distribution of binders. The Medpace OB-302 Team will attend the meeting. VIVUS will open the meeting and Medpace will present on the topics delegated by VIVUS. The meeting minutes will be prepared by Medpace, reviewed and approved by VIVUS, and distributed to the study sites by Medpace. The preparation of the meeting minutes is optional; however, is included in the budget. Medpace assumes the Investigators’ Meeting will serve as the initiation visit for the Investigators. Therefore, the budget reflects 20% of the sites will require an initiation visit (for those unable to attend the Investigators’ Meeting).
Clinical Trial Agreements for Sites, Central Laboratory, and EDC Vendor
Medpace will prepare and provide sample clinical trial agreements (including budgets) for the study sites. VIVUS will review and approve the final draft versions of the clinical trial agreements. The agreements will be distributed and negotiated with each site by Medpace (with final approval by VIVUS). Medpace will make payments to the clinical sites according to the VIVUS-approved schedule. All payments for sites, Medpace Reference Laboratories, and Phase
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Forward will be made electronically to Medpace within seven days of invoice receipt. If electronic payment exceeds or falls below actual costs, VIVUS will adjust based on the prior month’s payment reconciliation. Investigator payment invoices will include the following detail:
· Clinical study number;
· PI or Site #;
· Patient ID;
· Amounts paid per visit;
· Total amount earned to date;
· Prior payments; and
· Current payment amount.
Institutional Review Board and Initial Essential Documents Packages
Medpace will select a central Institutional Review Board (IRB) and coordinate the initial submissions to the IRB. VIVUS must approve the central IRB selected. Medpace will be responsible for payments to the central IRB utilizing funds provided in the same manner as described above.
A study-specific, prototype informed consent form (ICF) will be designed by Medpace. The ICFs will be reviewed and approved by VIVUS. Medpace will distribute the ICFs to the Central IRB. Medpace will be responsible for negotiating changes to the informed consents with the central IRB.
Deviations from the VIVUS template must be brought to the attention of the VIVUS Clinical Leader who will facilitate VIVUS legal review and approval, if required.
All components of the Initial Essential Documents Package will be collected, tracked, and maintained by Medpace according to Medpace SOPs. The Medpace Study Start-up team will review all documents, negotiate any changes with study site personnel, and correct any errors. The Initial Essential Document Package includes the following:
· Signed protocol signature pages;
· Financial disclosure questionnaires (FDQs) (template to be provided by VIVUS, Medpace to collect the forms);
· Clinical study agreement (includes study budget);
· FDA Form 1572;
· Laboratory certifications and reference values;
· Curricula vitae for all Investigators;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· IRB approval of the protocol (and any amendments), the informed consent and sponsor approved advertisements; and
· Qualification of IRB members.
The FDQs shall apply throughout the entire term of the study and for one year following last patient last visit (LPLV). If there is any change in the accuracy of a particular site’s FDQ during that time period, that site will be responsible for notifying Medpace of the change. Medpace will send a fax to all sites once the study has ended reminding them of their responsibilities (one of which includes notifying Medpace of any FDQ changes). If a site notifies Medpace of a change in staff from LPLV to one year after LPLV, Medpace will collect an updated FDQ and forward on to VIVUS. Costs associated with this task are included in the budget.
Site Initiation Visits
Site initiation visits will be conducted by the Medpace CRAs consistent with Medpace SOPs. For purposes of this proposal, it is assumed that the Investigators’ Meeting will serve as the initiation visit and only 20% of the sites for each study will require separate initiation visits. Typically, if the Investigator Meeting is considered the initiation visit, the CRA will contact the site via phone to review study procedures and the CRA’s first routine monitoring visit will occur shortly (can be defined as 1 or 2 weeks) after a patient is screened for the trial. During this visit, the CRA will review bullets 2-5 below. A site initiation visit report will be completed and forwarded to VIVUS within 10 business days of the visit. These visits will include, but are not limited to, the following tasks:
· Train site and applicable study personnel on the protocol and study procedures;
· Ensure the site has received all study supplies required for the conduct of the study, including study medication and access to eCRFs;
· Provide and review the Trial Master File binder. Medpace CRAs will provide instruction to the site personnel on the organization and maintenance of the documents in the binder;
· Review study medication accountability procedures;
· Provide eCRF completion instructions; and
· Explain the serious adverse event (SAE) reporting procedures.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
CLINICAL OPERATIONS
Monitoring Data Review Guidelines
The Medpace Lead CRA in collaboration with the project team members will develop a project-specific Monitoring Plan for the study. This plan will include detailed interpretations of study expectations for the CRAs assigned to the study. Issues are discussed and updated on an ongoing basis throughout the project. Medpace will request that VIVUS approve the initial document and then re-approve the document on a quarterly basis.
Routine Clinical Monitoring Visits
Medpace will conduct routine monitoring visits at each site consistent with Medpace SOPs. The frequency of the visits will be determined by the site’s activity, but will be conducted on average every four to six weeks. Visits at the beginning and end of the study may be more frequent based on the needs of the study, including, but not limited to, recruitment, quality data and study close-out activities. Based on recruitment being very rapid, the first routine visit will be performed within 2 weeks after the first two patients are screened at the site. The CRA will perform 100% source documentation. In addition, data queries will be resolved during the visits, eCRF changes will be verified, and supporting documentation for SAEs will be obtained. The Medpace CRA will verify all laboratory samples have been obtained according to guidelines and the results are available in the patient’s source documents. A monitoring visit report will be forwarded to VIVUS within 10 business days of the visit. VIVUS will be notified of any significant issues by phone within one business day.
The following tasks will also be performed:
· Train any new site personnel and review study issues with applicable site personnel;
· Ensure the site has sufficient study supplies (including study medication);
· Ensure the site is entering eligible patients into the study in a timely manner, and notify the Medpace Study Manager immediately of any problems;
· Detect any significant compliance or other issues and notify the Medpace Study Manager by phone, within one business day of the monitoring visit;
· Confirm the Trial Master File is complete and current, and the site is complying with applicable regulations and the protocol. VIVUS will be notified immediately of any significant deviations;
· Ensure the site is completing eCRFs in a timely manner;
· Ensure all completed eCRFs are reviewed, verified, corrected, and transmitted to Medpace;
· Review eCRFs for accuracy and protocol adherence;
· Verify study medication dispensing, compliance, and accountability for each patient; and
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Ensure the Investigator reported all SAEs to Medpace and the applicable IRB.
Medpace will provide a follow-up letter to the study site after each visit. The letter will include, but will not be limited to, the following:
· Important findings during the visit;
· Recommendations of corrective actions to be taken by the site; and
· Follow-up information regarding questions asked during the visit.
The Monitoring Visit Reports with all attachments including follow-up letters, will be available for view through the Medpace web based Clintrak® Study Management system within 10 days of the monitoring visit.
In-house Clinical Monitoring Activities
Investigators will be contacted on a regular basis (every week during the active recruitment period and between monitoring visits) to ensure progress at the study site. The CRA will take the opportunity to review enrollment, answer protocol-related questions, discuss eCRF completion issues, obtain information regarding AEs, and ensure the site continues to be committed to the completion of the study in a timely manner and according to the protocol.
Telephone contacts will be entered in the Medpace ClinTrak Study Management system. Contacts requiring urgent attention will be relayed to VIVUS immediately and will be resolved in collaboration with the Medpace CTM and the VIVUS Study Manager.
Withdrawals Due to Adverse Events
Withdrawals due to AEs will be tracked and reconciled with the eCRF database on an ongoing basis by Medpace. The CRA will be responsible for reporting withdrawals due to AEs to VIVUS using the monitoring visit report.
Medpace will write narratives for all withdrawals due to AEs, for use in the clinical trial study report.
Status Reporting
The Medpace Senior CTM will serve as the central channel for communication between Medpace and VIVUS. The OB-302 CTM will work in conjunction with the clinical monitoring group to track study progress and report to VIVUS on a weekly basis. In addition, the OB-302 CTM will be responsible for overall management of site information, overseeing the status of Investigator contracts, direct supervision of CRAs, tracking of enrollment information, and distribution of study supplies. The Medpace OB-302 CTM will be the primary contact for the sites to address protocol interpretations and inclusion/exclusion criteria. All protocol-related issues will be recorded in an ongoing document to ensure consistency. The CTM is available 24 hours a day, 7 days a week via the Medpace Project Helpline.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Medpace will develop a communication plan at the start-up of the study. The CTM will work with the VIVUS project team to define details regarding study communication, including status reporting and team conference calls. Medpace typically deliver status reports on a predetermined day (and time) of the week, which is established around the weekly team calls so that the information can be discussed. Utilization of IVRS will also allow the project team to review patient status on a real time basis.
The Medpace CTM will collaborate with the Medpace Medical Expert and the VIVUS Study Manager to address any questions that may arise. The ClinTrak Study Management databases will serve as the primary source of project status information and will allow the Medpace CTM to report on any aspect of the study. The databases are updated on a real-time basis, providing accurate and up-to-date information.
Elements of ClinTrak include:
· Phone contacts;
· Monitoring visit reports;
· Patient status including details on withdrawals during the treatment phase;
· Study supplies; and
· Protocol deviations.
Medpace will provide weekly status reports via a secure project website, to include the following status by site:
· Number of patients screened;
· Number of screen failures;
· Number of patients randomized;
· Number of patients dropped with drop rates; and
· Number of patients completed.
In addition, monthly reports will be provided, to include the following:
· Monitoring visits scheduled; and
· Monitoring visits completed.
Data Management status reports will be provided monthly via a secure website. These reports will include the following:
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Cumulative and interval eCRF status by site (including number of eCRFs transmitted and cleaned);
· Cumulative and interval patient status by site (including number of patients ongoing, completed, and early terminations) based on eCRF data in-house; and
· Cumulative and interval query status by site (including number of queries issued and days outstanding).
Project Website
Medpace will develop a secure OB-302 website that will be available to all project team members and site personnel. The website will include information and tools relevant to the study, such as status reports, meeting agendas and minutes, newsletters, monitor visit status, and the project timeline. Access is controlled by the type of user. Access to the tabs (sections) on the websites are controlled by the user type so that sites can have access to the section specifically designed for site access. Medpace can set up an automatic notification process of updates to the user email accounts. Clinical sites will have access only to parts of the website that pertain to their function. They will have the ability to receive study information and download study-related forms.
Team Meetings
VIVUS and Medpace
Medpace assumes that one face-to-face meeting, other than the Investigators’ meeting and kickoff meeting will take place for the OB-302 study at VIVUS.
Weekly teleconferences will be held during the study. The teleconferences will be held to discuss study progress and review project documents (as necessary). The Medpace Project Coordinators will be responsible for preparing and distributing agendas and minutes for each meeting/teleconference.
Additional meetings/teleconferences will be scheduled throughout the project, as needed.
Internal Meetings
Medpace will develop a project-specific internal project development/training program for all project team members. Included in this program will be the following:
· Protocol/eCRF review meeting;
· Medical in-services;
· Periodic Monitoring Plan meetings; and
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Periodic project meetings.
Newsletter
Medpace will prepare a 2-4 page full color site monthly newsletter for OB-302 as an additional avenue of communication and training for all site personnel. VIVUS will provide input and approval of the newsletter prior to distribution. Medpace will be responsible for printing and distributing two copies of each newsletter to each site.
Closeout Visits
Medpace will conduct closeout visits at each site consistent with Medpace SOPs after all patients have completed or discontinued from the study at the respective site. This visit may be performed as part of a final routine site monitoring visit. Site closeout visit reports will be forwarded to VIVUS within 10 business days of the visit. The following tasks will be performed:
· Resolve outstanding data queries;
· Ensure all study medication supplies are accounted for and that medication records and unused supplies are returned to VIVUS;
· Ensure the Investigator’s copies of data and source documents are properly stored;
· Ensure the Trial Master File is complete, correct, and properly stored;
· Ensure the Investigator is aware of record retention requirements and other obligations, and a final site status report is sent to the IRB and VIVUS; and
· Instruct the site to update the FDQs for one year after the study is completed.
Site Audits
Site audit visits will be conducted, as deemed necessary, by VIVUS.
Medpace will respond to any audit findings and ensure the proper actions are taken to resolve outstanding issues.
REGULATORY AFFAIRS AND SAFETY REPORTING
Serious Adverse Events
All SAEs will be reported immediately, within 24 hours of discovery or notification of the event, by the clinical study site to VIVUS, or its designee, according to SOPs specified by VIVUS.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
VIVUS will be responsible for submitting all immediately reportable SAEs (serious, causally related and unexpected) to the Food and Drug Administration (FDA) in accordance with the current regulations.
If a SAE has occurred at a site, the Medpace CRA will always 100% source document verify the event during the monitoring visit to ensure it has been recorded, documented, and reported appropriately and accurately. In addition, data queries will be resolved during the visits, CRF changes will be verified, and supporting documentation for SAEs will be obtained.
Medpace will provide VIVUS listings of non-serious adverse events from all sites to support filing of the Annual Safety Reports. Medpace will reconcile SAE listings with the AE database.
DATA MANAGEMENT
Data Management activities performed by Medpace will include eCRF tracking, preparation of a data management manual, eCRF review, coding of adverse events and concomitant medications, medical histories, data cleaning/editing, querying, query tracking, final database quality review, and delivery of the final SAS® database. The data cleaning process will be performed on an ongoing basis following Medpace Data Management SOPs. Two data transfers (SAS transport files) will be performed: a test transfer prior to FPFV and a final transfer. A unit price for additional data transfers has been provided in the budget.
Database Development and Data Management Manual
Medpace will design and validate the data entry systems prior to entry of data. A Data Management Manual will be prepared for each study using the Medpace template, and will include the following data management documents:
· Database specifications, based on VIVUS specifications;
· Guidelines for the tracking of eCRFs and data queries;
· Data Management Guidelines, which will include guidelines for reviewing the data, and description of the database edit check specifications to be performed for data cleaning; and
· Description of the database quality control (QC) plan.
The manuals will be reviewed and approved by VIVUS.
Data from the Central Laboratory
Medpace will arrange periodic data transfers from MRL. Medpace will track and reconcile discrepancies between the MRL demographic data and the eCRF database, which are generated during the data cleanup process throughout each project.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Data Entry and Data Querying
Medpace assumes no codable forms will be transmitted for screen failure patients. Data is entered by site personnel that have been trained on the eCRF system. Data will be reviewed according to Medpace Data Management Guidelines and edits. A data query will be generated electronically within the eCRF system. The resolutions/corrections are made by site personnel by changing the data. All changes are recorded in an audit trail. All answered queries are verified/closed by Medpace Data Management. All resolutions/corrections will be performed consistent with Medpace SOPs. The Data Coordinators will work directly with the site personnel in resolving queries.
Coding
Medpace will be responsible for coding adverse events, medical histories, and concomitant medications.
· MedDRA will be used to code adverse events and medical histories. Adverse events and medical histories will be coded to the lowest level term, preferred term, and system organ class.
· WHO DRUG will be used to code concomitant medications. Concomitant medications will be coded to the generic name and anatomic therapeutic class 3. It is assumed by Medpace that VIVUS holds a valid agreement with the Uppsala Monitoring Centre (UMC) for the WHO DRUG dictionary.
All coding will be done on a single version of each coding system (versions to be agreed upon by VIVUS). Medpace will provide the coding dictionaries.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Data Flow Chart
STATISTICAL ANALYSIS
Medpace will develop the data analysis plan (DAP) per the VIVUS/Medpace format and template. VIVUS will review and approve the DAP prior to initiation of programming. Included in the DAP is a detailed statistical methodology and programming specification for all statistical analyses, tables/figures/listings (TFLs), and derived datasets.
Medpace will be responsible for programming and generating all TFLs, according to the Medpace standard analysis validation and quality control procedures. All TFLs will be programmed using SAS (Version 8), according to the VIVUS Programming Standards document. Pre-final TFLs will be generated twice on clean data for format review. Final TFLs will be generated on final data for the clinical study report.
MEDICAL WRITING
The Medpace Medical Writing team works in collaboration with the Medpace Medical Expert to provide a clinical study report according to FDA/ICH guidelines. The preparation of the Integrated Clinical/Statistical Study Report involves three stages of development: (1) the Study Report Shell (SRS), (2) the Pre-Final Study Report (PFSR), and (3) the Final Study Report (FSR).
The SRS is prepared after sign-off of the Final DAP. The SRS is created using a template and/or style guide provided by VIVUS, or by utilizing the Medpace standard report template, which
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
adheres to the ICH guideline, “Structure and Content of Clinical Study Reports,” and follows the American Medical Association Manual of Style. The SRS incorporates information from the protocol, amendments, and eCRFs into sections of the report, including but not limited to the study design, study population, treatments administered, and the evaluation schedule. The statistical methods and results sections encompass information derived from the Final DAP. The results sections include mock-up in-text tables and text. The SRS undergoes a complete team review, which includes the Medical Monitor, Statistician, CTM, Data Manager, and other team members, if applicable. After the Medical Writer incorporates all team changes into the SRS, the SRS undergoes Medpace’s comprehensive document QC process. Once all changes from the document QC process are implemented into the SRS, the SRS is forwarded to the VIVUS for review.
Preparation of the PFSR occurs after receipt and implementation of VIVUS comments on the SRS, declaration of a clean database, and completion of Pre-Final Analyses. If necessary, a results review meeting is conducted with key members of the Medpace and VIVUS project team as the Medical Writer begins preparing the PFSR. The PFSR is a complete version of the report without the appendices. The PFSR undergoes a complete team review, which includes the Medical Monitor, Statistician, CTM, Data Manager, and other team members, if applicable. After the Medical Writer incorporates all team changes into the PFSR, the PFSR undergoes Medpace’s comprehensive document QC process. Once all changes from the document QC process are implemented into the PFSR, the PFSR is forwarded to VIVUS for review.
The FSR is prepared once VIVUS’ comments on the PFSR are returned and all requested changes are agreed upon (including the acceptance of final text for all complicated or sensitive sections, which may require sending non-QC’d drafts of the report to VIVUS), and the Final Analyses are completed. The FSR is a complete version of the report including paginated appendices and/or supplements. The FSR undergoes a complete internal QC review and is forwarded to VIVUS for sign-off. The signed cover sheet is returned by VIVUS to verify their acceptance of the FSR.
STUDY CLOSEOUT
At the conclusion of each study, once all deliverables have been met, Medpace will return the original study files to VIVUS. These will include:
· General project administration files (this file includes items such as: Outside Vendor Correspondence, Multiple Site Correspondence (e.g. faxes to all sites), Central IRB Correspondence, Project Specific SOPs and Procedures, Monitoring Plan, Trial Master File, Project Timelines, Newsletters, and Meeting Minutes)
· Site files (this file includes site items such as: Essential Documents, Budget, Site Correspondence, Monitoring Visit Reports, Drug shipment documents, Study Supply forms, and Protocol Deviations);
· Final statistical tables and listings with results;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Data management documentation (this file includes items such as: Database Definition Document, Final eCRFs, External Database Import Specs., Data Management Plan, Coding Reports, Analysis Plan, and Randomization Code); and
· Final clinical study report.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Assumptions
|
OB-302 |
|
Description |
|
Number of Investigators |
|
*** |
|
Number of Back-up Investigators |
|
*** |
|
Number of Screened Patients |
|
*** |
|
Number of Randomized Patients |
|
*** |
|
Number of Randomized Patients/Site |
|
*** |
|
Duration of Enrollment Period |
|
*** |
|
Duration of Treatment Period |
|
*** |
|
Number of Investigators’ Meetings |
|
*** |
|
Number of Kickoff Meetings (at Medpace) |
|
*** |
|
Number of Sponsor Meetings (at VIVUS) |
|
*** |
|
Number of Conference Calls |
|
*** |
|
Frequency of Conference Calls |
|
*** |
|
Number of Clinical Monitors |
|
*** |
|
Number of Pre-study Visits |
|
*** |
|
Number of Initiation Visits |
|
*** |
|
Number of Routine Monitoring Visits |
|
*** |
|
Number of Closeout Visits |
|
*** |
|
Monitoring Frequency |
|
*** |
|
Number of Newsletters per Site (monthly) |
|
*** |
|
Estimated Number of eCRFs per Completed Patient |
|
*** |
|
Estimated Number of Unique eCRFs per Completed Patient |
|
*** |
|
Total Number of eCRFs |
|
*** |
|
Estimated Number of AE Codes Per Patient |
|
*** |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
Estimated Number of Medical History Codes Per Patient |
|
*** |
|
Estimated Number of Concomitant Medications Codes Per Patient |
|
*** |
|
Estimated Number of Queries Per Patient |
|
*** |
|
External Data Sources |
|
*** |
|
Data Transfers from Medpace to VIVUS |
|
*** |
|
Number of Raw Listings |
|
*** |
|
Number of Unique TFs |
|
*** |
|
Number of Version TFs |
|
*** |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #02
Appendix 3 — Project Schedule
VIVUS, Inc.
Qnexa ▇▇-▇▇▇
▇▇▇▇▇▇▇▇▇▇
|
▇▇-▇▇▇ |
|
Date |
|
Medpace Begins Work |
|
*** |
|
Protocol Finalized |
|
*** |
|
First Patient First Visit |
|
*** |
|
Last Patient First Visit |
|
*** |
|
Last Patient Last Visit |
|
*** |
|
Final Database Lock |
|
*** |
|
Final TFLs Available |
|
*** |
|
Delivery of Final Clinical Study Report |
|
*** |
|
CONFIDENTIAL |
September 11, 2007 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #02
Appendix 4 — Budget
VIVUS, Inc.
Qnexa OB-302
Medpace Fee Estimate
***
|
CONFIDENTIAL |
September 11, 2007 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #02
Appendix 5 — Payment Schedule
VIVUS, Inc.
Qnexa OB-302
Payment Schedule
As set forth in this Agreement, professional service fees totalling $*** will be paid by VIVUS for professional services rendered by Medpace according to the following schedule.
***
Pass-through expenses will be billed to VIVUS on a monthly basis as incurred. The first Wire-Transfer payment will be invoiced prior to any site becoming active to ensure funds are available for site payments. Medpace will pay sites immediately following receipt of Wire-Transfers. Medpace will invoice VIVUS on a monthly basis and payments will be made by VIVUS within seven days of invoice receipt.
Payment Information and General Conditions
***
Inflation
The fees stipulated in the fee estimate include inflation for the duration of the study as specified in this proposal. Any significant shift in timelines will require a revision to the fees.
|
CONFIDENTIAL |
September 11, 2007 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
Appendix 6 |
|
Transfer of Obligations Form |
|
CONFIDENTIAL |
|
|
Directions: Complete a form for each clinical study where Sponsor obligations have been transferred in accordance with 21 CFR Part 312, Subpart D (Responsibilities of Sponsors). Forward the completed form to Sponsor’s Regulatory Affairs Department for submission to the applicable regulatory agencies.
|
Drug: |
|
VI-0521 |
Study ID: OB 302 | |
|
|
|
|
| |
|
Study Title: |
|
A phase III randomized, double blind, placebo controlled multicenter study to determine the safety and efficacy of VI-0521 in the treatment of obesity in otherwise healthy adults. | ||
|
|
|
| ||
|
CRO Name: |
|
Medpace, Inc. | ||
|
|
|
| ||
|
CRO Address: |
|
*** | ||
OBLIGATIONS TRANSFERRED TO MEDPACE: x THE APPROPRIATE BOX(ES).
o All obligations in 21 CFR 312, Subpart D (Responsibilities of Sponsors) have been transferred to Medpace.
x The following obligations have been transferred to Medpace:
Sec. 312.32: IND Safety Reports
x Promptly review safety information. *Sponsor will be notified within one (1) business day of discovery of significant new or serious adverse events or risks, or any unusual frequency of reactions with respect to the drug.
o Notify all participating investigators in a written IND safety report of any AE associated with the drug that is both serious and unexpected.
o Notify the FDA in a written IND safety report of any AE associated with the drug that is both serious and unexpected.
Sec. 312.53: Selecting investigators and monitors
x (a) Select qualified investigators
x (b) Control investigational drug shipment
x (c) Obtain information from investigators
x (1) Signed Form FDA-1572
x (2) CV or other qualification statement
x (3) Clinical protocol outline
x (4) Financial disclosure information
x (d) Select qualified monitors
Sec. 312.54: Emergency research
o (a) Monitor the progress of all studies involving an exception from informed consent.
o (b) Monitor such studies to identify when an IRB determines that it can’t approve the research.
Sec. 312.55: Informing investigators
x (a) Provide sites with the current Inv. Brochure.
x (b) Inform investigators of new observations on the drug, particularly with respect to AEs and safe use.
Sec. 312.56: Review of ongoing investigations
o (a) Monitor the progress of all IND studies.
x (b) Secure compliance from noncompliant investigators or discontinue drug shipments and end the investigator’s participation in the study.
o (c) Review and evaluate the safety and efficacy results as it is obtained from the investigator.
x (d) Discontinue use of the investigational drug if it is determined to present an unreasonable and significant risk to subjects, notify all IRBs and investigators, and assure the return or alternate disposition of the drug from the investigators.
Sec. 312.57: Record keeping and record retention
x (a) Maintain adequate records showing investigational drug receipt, shipment, or other disposition. *Master Drug Logs will include the name of the Investigator to whom the drug is shipped, the date, and the quantity and batch of each such shipment.
x (b) Maintain complete and accurate records showing any financial interests of the investigator subject to 21 CFR 54.
x (c) Retain the records and reports required by the regulations for 2 years after the marketing application is approved, or if not approved, until 2 years after investigational drug shipment is discontinued and FDA has been notified.
o (d) Retain reserve samples of any test article and reference standard identified and used in bioequivalence or bioavailability studies.
Sec. 312.58: Inspection of sponsor’s records and reports
x (a) Permit FDA personnel to have access to and copy and verify any records and reports related to the clinical investigation.
x (b) Permit DEA personnel to have access to and copy records related to the shipment, delivery, receipt and disposition of any investigational controlled substance. Assure adequate storage precautions are taken for investigational new drug substances listed in any schedule of the Controlled Substances Act.
Sec. 312.59: Disposition of unused supply of investigational drug
x Assure the return (or alternate disposition) of all unused supplies of the investigational drug from each discontinued/terminated investigator; maintain written records of any disposition of the investigational drug.
Other
o Please describe any other applicable transfers below:
September 7, 2007
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
MEDPACE
VIVUS Task Order #03
EXHIBIT A
Qnexa OB-303 TASK ORDER
MEDPACE Task Order Number: 03
MEDPACE Project Number: VOB303
This Task Order, dated September 12, 2007, is between Medpace, Inc. (“MEDPACE”), and VIVUS, Inc. (“VIVUS”).
RECITALS:
WHEREAS, MEDPACE and VIVUS have entered into that certain Master Services Agreement dated September 12, 2007 (the “Master Services Agreement”); and
WHEREAS, pursuant to the Master Services Agreement, MEDPACE has agreed to perform certain Services in accordance with Task Orders from time to time entered into by the Parties and VIVUS and MEDPACE now desire to enter into such a Task Order; and
WHEREAS, MEDPACE and VIVUS desire that MEDPACE provide certain services with respect to a phase III randomized, double blind, placebo controlled multicenter study to determine the safety and efficacy of VI-0521 in the treatment of obesity in adults with obesity-related comorbid conditions (the “Study”) for the study of the product VI-0521 (“Study Product”) as set out in the Protocol Number: OB-303, which is attached hereto as Appendix 1;
NOW, THEREFORE, in consideration of the mutual covenants contained herein, the Parties hereby agree as follows:
1. Scope of Work: MEDPACE shall perform the services described in the Scope of Work, attached hereto as Appendix 2, in accordance with the Project Schedule, attached hereto as Appendix 3 and any other documents attached to and specifically referenced in this Task Order (“Services”)
2. Compensation: For performance of these Services, VIVUS shall pay to MEDPACE an amount equal to the Project Budget set forth in Appendix 4, which amount shall be payable pursuant to the Payment Schedule set forth in Appendix 5.
3. Transfer of Obligations: Sponsor Obligations transferred to MEDPACE by VIVUS (consistent with the regulations set forth in 21 C.F.R. Section 312, Subpart D) are identified in Appendix 6.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
4. MSA. The provisions of the Master Services Agreement are hereby expressly incorporated by reference into and made a part of this Task Order.
IN WITNESS WHEREOF, the Parties have hereunto signed this Task Order effective as of the day and year first written above.
|
MEDPACE, INC. |
| ||
|
|
| ||
|
Signature: |
/s/ August ▇. ▇▇▇▇▇▇▇▇ |
| |
|
|
|
| |
|
By: |
August ▇. ▇▇▇▇▇▇▇▇ |
| |
|
(Print Name) |
| ||
|
|
|
| |
|
Title: |
President |
| |
|
|
|
| |
|
Date: |
September 12, 2007 |
| |
|
|
| ||
|
|
| ||
|
SPONSOR |
| ||
|
|
| ||
|
Signature: |
/s/ ▇▇▇▇▇▇ ▇. Day |
| |
|
|
|
| |
|
By: |
▇▇▇▇▇▇ ▇. Day |
| |
|
(Print Name) |
| ||
|
|
|
| |
|
Title: |
Vice President, Clinical Development |
| |
|
|
|
| |
|
Date: |
September 10, 2007 |
| |
List of Appendices:
Appendix 1: Protocol
Appendix 2: Scope of Work
Appendix 3: Project Schedule
Appendix 4: Project Budget
Appendix 5: Payment Schedule
Appendix 6: Transfer of Obligations
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
VI-0521
Protocol No. OB-303
***
CLINICAL PROTOCOL
A PHASE III RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED MULTICENTER STUDY TO DETERMINE THE SAFETY AND EFFICACY OF VI-0521 IN THE TREATMENT OF OBESITY IN ADULTS WITH OBESITY-RELATED CO-MORBID CONDITIONS
|
Compound: |
VI-0521 |
|
|
|
|
Compound Name (if applicable): |
Phentermine plus Topiramate |
|
|
|
|
US IND Number (if applicable): |
*** |
|
|
|
|
Protocol Number: |
OB 303 |
|
|
|
|
Phase: |
3 |
|
|
|
|
Medical Monitor: |
*** |
|
|
|
|
Sponsor: |
VIVUS, Inc. |
|
|
|
|
Version and Date: |
*** |
This document contains confidential information belonging to VIVUS. Except as otherwise agreed to in writing, by accepting or reviewing this document, you agree to hold this information in confidence and not copy or disclose it to others (except where required by applicable law) or use it for unauthorized purposes. In the event of any actual or suspected breach of this obligation, VIVUS must be promptly notified.
VIVUS — Company Confidential
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
INTERNAL PROTOCOL APPROVAL
Protocol Number: OB-303
Title: A Phase III Randomized, Double-Blind, Placebo Controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in Adults with Obesity-Related Co-Morbid Conditions
The signature below documents that the reviewer has read and approved the attached protocol.
|
|
|
Signature |
|
Date |
|
|
|
|
|
|
|
Author: ▇▇▇▇▇▇▇▇ ▇▇▇▇▇▇, PhD, MPH |
|
|
|
|
|
Clinical Consultant |
|
|
|
|
|
|
|
|
|
|
|
▇▇▇▇▇▇ Day, PhD |
|
|
|
|
|
VP, Clinical Development |
|
|
|
|
|
|
|
|
|
|
|
▇▇▇▇▇▇▇▇▇▇ ▇▇▇▇▇▇▇▇▇, PhD |
|
|
|
|
|
Sr. Director, Regulatory Affairs |
|
|
|
|
|
|
|
|
|
|
|
▇▇▇ ▇▇▇▇▇▇ |
|
|
|
|
|
▇▇. Director, Pharmaceutical Development |
|
|
|
|
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
PRINCIPAL INVESTIGATOR SIGNATURE
Protocol Number: OB-303
Title: A Phase III Randomized, Double-Blind, Placebo Controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in Adults with Obesity-Related Co-Morbid Conditions
The signature below indicates that the principal investigator has read and understands the protocol and agrees to conduct the study in accordance with the protocol, applicable guidelines for Good Clinical Practices, the Declaration of Helsinki and all applicable regulatory guidelines and requirements. Please return one copy of this executed page to VIVUS, Inc.
|
Printed Name: |
|
|
|
|
|
Signature: |
Date: |
|
|
|
|
|
|
|
Facility Name: |
|
|
|
|
|
Address: |
|
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
PROTOCOL SYNOPSIS
Rationale:
Obesity leads to the development of co-morbidities such as hypertension, type 2 diabetes, dyslipidemia, coronary artery disease (CAD) and stroke. Weight reduction in obese individuals has been shown to delay or prevent the onset of these co-morbidities, or even reverse the damage caused by these co-morbidities. Diet, exercise and behavior modification therapy can be effective short-term treatments; however, many people experience difficulty in achieving and maintaining weight reduction without pharmacotherapy. VI-0521 is an investigational weight loss therapy that is a new combination of two currently approved drugs, phentermine and topiramate. This novel combination treatment may provide a safe and effective option for the achievement and maintenance of weight loss in obese adults.
Objectives:
The objectives of this study are to evaluate the safety and efficacy of two doses of VI-0521 compared to placebo in the treatment of overweight and obesity in adults with at least *** obesity-related co-morbid conditions.
Trial Design:
The study is a randomized, double blind placebo-controlled prospective trial with subjects randomized to receive daily treatment with VI-0521 *** or *** or ***, with the total duration of treatment being 56 weeks. Randomization will be stratified by ***, and at least *** of the subjects must be ***. Approximately 2500 subjects will be treated under the protocol with *** subjects randomized to ***, and ***. Up to *** study sites in the USA will be employed.
At randomization, subjects will be instructed to follow a hypocaloric diet representing a 500-calorie/day deficit and advised to implement a lifestyle modification program, as tolerated, throughout the study period. During the first 4 weeks of treatment (weeks 1-4), study medication will be titrated to the desired level, with the dosage level increased each week as determined by randomization group. During treatment weeks 5-56, the dose will be maintained at the final dose level. Subjects who are unable to tolerate the assigned dosage may be treated at a reduced dose level and/or interrupt dosing for up to ***.
Subjects will return at 4-week intervals for measurement and evaluation. Female subjects of child bearing potential will undergo a urine pregnancy test at each study visit. Subjects who discontinue the study during treatment will be encouraged to return at the 56-week time point for evaluation.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Study Subjects:
The study population will consist of *** (BMI > *** and < ***) adults < 70 years of age with at least *** of the following obesity-related comorbid conditions:
· ***;
· ***;
· ***;
· ***.
Major exclusion criteria include: type 1 diabetes; diabetes medications other than metformin; known or suspected valvular heart disease; clinically significant ECG abnormality, physical exam, ▇▇▇▇▇ ▇▇▇▇▇ or laboratory abnormality; clinically significant hepatic or renal disease; creatinine clearance < 60 mL/minute; clinically significant thyroid dysfunction as evidenced by signs, symptoms, or TSH > 1.5 x ULN; obesity of known genetic or endocrine origin; history of bipolar disorder or psychosis, depression of moderate or greater severity, or presence or history of suicidal behavior or active suicidal ideation; recent weight instability; history of glaucoma or increased intraocular pressure; prior bariatric surgery; or smoking cessation within 3 months prior to enrollment. Female subjects of childbearing potential must agree to use adequate contraception (a double barrier method, stable hormonal contraception plus single barrier or tubal ligation) for the duration of treatment.
Efficacy Endpoints:
The primary endpoints are the differences between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), and percent of subjects with weight loss of 5% or more at end of treatment (week 56). Percent weight loss will be calculated as ***.
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) for *** and *** groups;
· The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups; and
· The difference in change in waist circumference (randomization to week 56) for *** and *** groups.
Additional efficacy endpoints include:
· Effect on ***;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Effect on body composition as indicated by ***. *** assessments will be performed at only at a ***.
· Effect on *** of *** and ***;
· Change from baseline to week 28 and week 56 in BMI;
· The change in obesity-associated risk factors (HgbA1c, total cholesterol, triglycerides, LDL-C, HDL-C, fasting glucose, fasting insulin, a measure of insulin sensitivity, systolic blood pressure, diastolic blood pressure, C-reactive protein);
· Change in urinary microalbumin and albumin/creatinine ratio (ACR) from screening to week 28 and week 56;
· Change from baseline in medication number and dosages for medication to treat cardiovascular or metabolic risk factors;
· Difference between *** and *** groups in the rate of progression to type 2 diabetes (subjects non-diabetic at screening); and
· Baseline adjusted change in Framingham 10-year risk score at weeks 28 and 56.
The change in weight loss (absolute, percent, percent of subjects achieving weight loss of > 5% and > 10% of starting weight), waist circumference and obesity associated risk factors will also be assessed over time. Subgroup analyses, including but not limited to analysis by gender, age and race, may be performed.
*** Assessment:
*** will also be evaluated. Data will be obtained using a multiple trough sampling scheme with samples collected at *** and ***. Effects of various cofactors including (but not limited to) ***, gender, race, *** and age will be evaluated.
Safety Endpoints:
Safety will be assessed by an evaluation of adverse events, including eye symptoms (collected at each study visit); ***. All subjects will be screened for the *** using a validated survey instrument ***, and for *** using the ***. Follow-up *** assessments will be done at *** after ***.
Statistical Methods:
All subjects who are randomized, take one or more doses of test material and have at least one post treatment efficacy measurement will be included in the analysis. Comparisons between treatments will be assessed using an *** with factors of ***, *** and *** and with *** for percent weight loss, and *** for percentage of subjects with at least 5% weight loss. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant at *** for
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
both co-primary endpoints, then the test will proceed to the *** also at the ***. If the statistical comparison is not significant at the ***, then the statistical test will be stopped and the *** will not be tested. If both dose groups are significantly better than ***, then the two active dose groups will be compared. A *** of difference in response rate between treatment groups will be derived. The *** for subjects who discontinue treatment prior to completion of the study, ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
TABLE OF CONTENTS
|
INTERNAL PROTOCOL APPROVAL |
2 |
|
|
|
|
PRINCIPAL INVESTIGATOR SIGNATURE |
3 |
|
|
|
|
PROTOCOL SYNOPSIS |
4 |
|
|
|
|
1. INTRODUCTION |
15 |
|
|
|
|
1.1. Background |
15 |
|
|
|
|
1.2. Rationale |
16 |
|
|
|
|
2. TRIAL OBJECTIVES |
16 |
|
|
|
|
3. TRIAL DESIGN |
17 |
|
|
|
|
4. SUBJECT SELECTION |
18 |
|
|
|
|
4.1. Inclusion Criteria |
18 |
|
|
|
|
4.2. Exclusion Criteria |
19 |
|
|
|
|
4.3. Randomization Criteria |
21 |
|
|
|
|
4.4. Life Style Guidelines |
21 |
|
|
|
|
5. TRIAL TREATMENTS |
22 |
|
|
|
|
5.1. Allocation to Treatment |
22 |
|
|
|
|
5.2. Breaking the Blind |
22 |
|
|
|
|
5.3. Drug Supplies |
22 |
|
|
|
|
5.3.1. Formulation and Packaging |
22 |
|
|
|
|
5.3.2. Preparation and Dispensing |
23 |
|
|
|
|
5.3.3. Administration |
23 |
|
|
|
|
5.3.4. *** |
23 |
|
|
|
|
5.3.5. Compliance |
24 |
|
|
|
|
5.4. Drug Storage and Drug Accountability |
24 |
|
|
|
|
5.5. Concomitant Medication(s) |
24 |
|
|
|
|
5.5.1. Excluded Medications |
24 |
|
|
|
|
5.5.2. Other Restricted Medications |
25 |
|
|
|
|
5.5.3. Documentation of Concomitant Medication Use |
25 |
|
|
|
|
5.6. Treatment of Diabetes |
25 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
5.7. Treatment of *** |
26 |
|
|
|
|
5.8. Treatment of *** |
26 |
|
|
|
|
6. TRIAL PROCEDURES |
26 |
|
|
|
|
6.1. Screening |
27 |
|
|
|
|
6.1.1. Visit 1 |
27 |
|
|
|
|
6.1.2. Visit 1a |
28 |
|
|
|
|
6.2. Trial Period |
28 |
|
|
|
|
6.2.1. Randomization (Visit 2) |
28 |
|
|
|
|
6.2.2. Titration (Visit 3) |
29 |
|
|
|
|
6.2.3. Treatment (Visits 4 through 16) |
29 |
|
|
|
|
6.2.4. End of Treatment (Visit 17 or Withdrawal from Study) |
30 |
|
|
|
|
6.3. Study Period |
31 |
|
|
|
|
6.4. Subject Withdrawal |
31 |
|
|
|
|
7. ASSESSMENTS |
32 |
|
|
|
|
7.1. Weight Assessment and Waist Measurement |
32 |
|
|
|
|
7.1.1. Weight Assessment |
33 |
|
|
|
|
7.1.2. Waist Measurement |
33 |
|
|
|
|
7.1.3. Height and BMI |
34 |
|
|
|
|
7.2. ▇▇▇▇▇ ▇▇▇▇▇ |
34 |
|
|
|
|
7.3. Questionnaires |
34 |
|
|
|
|
7.3.1. *** |
34 |
|
|
|
|
7.3.2. *** |
34 |
|
|
|
|
7.3.3. *** |
35 |
|
|
|
|
7.3.4. *** |
35 |
|
|
|
|
7.4. *** and End of Treatment Questions |
35 |
|
|
|
|
7.4.1. *** |
35 |
|
|
|
|
7.4.2. End of Treatment Questions |
35 |
|
|
|
|
7.5. Laboratory Tests |
35 |
|
|
|
|
7.5.1. Blood Chemistry |
36 |
|
|
|
|
7.5.2. Oral Glucose Tolerance Test |
36 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
7.5.3. Hematology |
36 |
|
|
|
|
7.5.4. Urinalysis |
36 |
|
|
|
|
7.5.5. Biomarkers |
36 |
|
|
|
|
7.5.6. Hemoglobin A1c |
37 |
|
|
|
|
7.5.7. Urine Pregnancy Test |
37 |
|
|
|
|
7.5.8. Thyroid Stimulating Hormone (TSH) |
37 |
|
|
|
|
7.5.9. Antibody to HIV, HCV, HBsAg |
37 |
|
|
|
|
7.5.10. C-Peptide |
37 |
|
|
|
|
7.5.11. Screening Serum Sample (Retain) |
37 |
|
|
|
|
7.5.12. Urine Drug Screen |
38 |
|
|
|
|
7.6. *** Evaluation |
38 |
|
|
|
|
7.7. Physical Examination |
38 |
|
|
|
|
7.8. Electrocardiogram (ECG) |
38 |
|
|
|
|
7.9. Framingham Risk Score |
38 |
|
|
|
|
7.10. Body Composition |
39 |
|
|
|
|
8. ADVERSE EVENT REPORTING |
39 |
|
|
|
|
8.1. Adverse Events |
39 |
|
|
|
|
8.1.1. Severity Assessment |
39 |
|
|
|
|
8.1.2. Causality Assessment |
40 |
|
|
|
|
8.1.3. Abnormal Test Findings |
40 |
|
|
|
|
8.2. Serious Adverse Events |
40 |
|
|
|
|
8.2.1. Definition of Hospitalization |
41 |
|
|
|
|
8.3. Eliciting Adverse Event Information |
41 |
|
|
|
|
8.3.1. Eye Pain |
42 |
|
|
|
|
8.3.2. *** |
42 |
|
|
|
|
8.3.3. Hypoglycemia |
42 |
|
|
|
|
8.4. Reporting Period |
42 |
|
|
|
|
8.5. Reporting Requirements |
43 |
|
|
|
|
8.5.1. Serious Adverse Event Reporting Requirements |
43 |
|
|
|
|
8.5.2. Non-Serious Adverse Event Reporting Requirements |
43 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
8.5.3. *** |
43 |
|
|
|
|
9. DATA ANALYSIS/STATISTICAL METHODS |
44 |
|
|
|
|
9.1. Sample Size Determination |
44 |
|
|
|
|
9.2. Efficacy Analysis |
44 |
|
|
|
|
9.2.1. Analysis of Primary Endpoints |
44 |
|
|
|
|
9.2.2. Analysis of Secondary Endpoints |
45 |
|
|
|
|
9.3. Analysis of Other Endpoints |
45 |
|
|
|
|
9.3.1. Other Efficacy Endpoints |
45 |
|
|
|
|
9.3.2. *** Analysis |
46 |
|
|
|
|
9.4. Analysis Populations |
46 |
|
|
|
|
9.5. *** |
46 |
|
|
|
|
9.6. *** |
46 |
|
|
|
|
9.7. Safety Analysis |
46 |
|
|
|
|
9.7.1. Adverse Events |
46 |
|
|
|
|
9.7.2. Clinical Laboratory Tests |
47 |
|
|
|
|
9.7.3. ▇▇▇▇▇ ▇▇▇▇▇ and Other Safety Evaluations |
47 |
|
|
|
|
9.7.4. Questionnaire Assessments |
47 |
|
|
|
|
9.8. Interim Analysis |
47 |
|
|
|
|
9.9. *** |
48 |
|
|
|
|
10. QUALITY CONTROL AND QUALITY ASSURANCE |
48 |
|
|
|
|
11. DATA HANDLING AND RECORD KEEPING |
48 |
|
|
|
|
11.1. Case Report Forms / Electronic Data Record |
48 |
|
|
|
|
11.2. Record Retention |
49 |
|
|
|
|
12. ETHICS |
49 |
|
|
|
|
12.1. Institutional Review Board (IRB)/Independent Ethics Committee (IEC) |
49 |
|
|
|
|
12.2. Ethical Conduct of the Trial |
50 |
|
|
|
|
12.3. Subject Information and Consent |
50 |
|
|
|
|
12.4. Disclosure of Data |
50 |
|
|
|
|
13. REGULATORY CORRESPONDENCE |
50 |
|
|
|
|
14. DEFINITION OF END OF TRIAL |
51 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
15. SPONSOR DISCONTINUATION CRITERIA |
51 |
|
|
|
|
16. PUBLICATION OF TRIAL RESULTS |
51 |
|
|
|
|
17. REFERENCES |
53 |
|
|
|
|
APPENDIX 1: SCHEDULE OF STUDY ACTIVITIES |
56 |
|
|
|
|
APPENDIX 2: *** |
57 |
|
|
|
|
APPENDIX 3: *** |
60 |
|
|
|
|
APPENDIX 4: *** |
64 |
|
|
|
|
APPENDIX 5: PROTOCOL AMENDMENTS |
68 |
|
|
|
|
TABLES |
|
|
|
|
|
Table 1: VI-0521 Dosage Strengths by Titration Week for Each Treatment Group |
23 |
|
|
|
|
Table 2. Guidelines for adverse event reports based on *** |
42 |
|
|
|
|
FIGURES |
|
|
|
|
|
Figure 1. Schematic Representation of Study Design |
17 |
|
|
|
|
Figure 2. Measuring Tape Position for Waist Circumference Assessments |
33 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
LIST OF ABBREVIATIONS
|
ACE |
angiotensin converting enzyme |
|
ACR |
albumin/creatinine ratio |
|
*** |
*** |
|
*** |
*** |
|
*** |
*** |
|
CAD |
coronary artery disease |
|
Cl |
apparent clearance |
|
cm |
centimeter |
|
CRFs |
case report forms |
|
*** |
*** |
|
*** |
*** |
|
DSM-IV |
Diagnostic and Statistical Manual IV |
|
ECG |
electrocardiogram |
|
FDA |
Food and Drug Administration |
|
*** |
*** |
|
GCP |
Good Clinical Practices |
|
*** |
*** |
|
gm |
gram |
|
HBsAg |
hepatitis B surface antigen |
|
HCV |
hepatitis C virus |
|
HDL-C |
high density lipoprotein cholesterol |
|
HIV |
human immunodeficiency virus |
|
I-CAM |
Intercellular Cell Adhesion Molecule |
|
ICH |
International Conference on Harmonization |
|
IEC |
Independent Ethics Committee |
|
IND |
Investigational New Drug |
|
IRB |
Institutional Review Board |
|
IVRS |
Interactive Voice Response System |
|
*** |
*** |
|
kcal/day |
kilocalories per day |
|
kg |
kilogram |
|
kg/m2 |
kilogram per square meter |
|
*** |
*** |
|
LDL-C |
low density lipoprotein cholesterol |
|
µU/mL |
microunits per milliliter |
|
MedDRA |
Medical Dictionary for Regulatory Activities |
|
mg |
milligram |
|
mg/day |
milligram per day |
|
mg/dL |
milligram per deciliter |
|
mmHg |
millimeters of mercury |
|
NYHA |
New York Heart Association |
|
OGTT |
oral glucose tolerance test |
|
PAI-1 |
Plasminogen Activator Inhibitor 1 |
|
*** |
*** |
|
*** |
*** |
|
QOD |
every other day |
|
RBC |
red blood cell |
|
RBP-4 |
Retinol Binding Protein 4 |
|
*** |
*** |
|
*** |
*** |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
TSH |
thyroid stimulating hormone |
|
ULN |
upper limit of normal |
|
*** |
*** |
|
V-CAM |
Vascular Cell Adhesion Molecule |
|
WBC |
white blood cells |
|
WHO |
World Health Organization |
|
WNL |
within normal limits |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
1. INTRODUCTION
VI-0521 is an investigational weight loss therapy that is a combination of two approved drugs, phentermine and topiramate. Phentermine hydrochloride (phentermine), an anorectic agent, has sympathomimetic actions and stimulates the central nervous system.(1), (2) It is postulated that ***, and there is a ***.(3), (4) Topiramate, an anticonvulsant agent that has been shown to induce weight loss, is known to ***.(5) However, the specific mechanism mediating the weight loss is not known. The present study is being conducted to evaluate the combined use of phentermine and topiramate at doses of 7.5 mg phentermine/46 mg topiramate and 15 mg phentermine/92 mg topiramate in the treatment of obesity in adult subjects.
1.1. Background
Recent studies have shown that about 129 million adults in the United States are clinically overweight or obese.(6) In recent years, there has been a dramatic increase in obesity in both children and adults.(7), (8) Results from the National Health and Nutrition Examination Survey showed an overall 32.2% prevalence of obesity during 2003-2004, with obesity increasing from 27.5% in 1999-2000 to 31.1% in 2003-2004 in men but remaining relatively unchanged in women (1999-2000, 33.4%; 2003-2004, 33.2%).(8)
Obesity is associated with numerous co-morbidities including dyslipidemia, coronary artery disease (CAD), hypertension, stroke and type 2 diabetes.(7), (9) Epidemiological data indicate that obesity is associated with increased mortality,(10) and a recent study of over 500,000 individuals concluded that excess body weight during midlife, was associated with an increased risk of death.(11) A modest weight loss (5-10%) can result in a marked reduction in obesity-related metabolic and cardiovascular risk factors.(12), (13), (14) Diet, exercise and behavior modification are standard treatments for obesity although most obese individuals do not achieve prolonged weight reduction without supplemental pharmacotherapy. However, the medications currently approved by the Food and Drug Administration (FDA) for weight loss are often poorly tolerated due to side effects and often fail to maintain long-term efficacy.(15)
Phentermine hydrochloride, a synthetic sympathomimetic amine, is an anorectic agent approved by the FDA as a short-term adjunct to a weight loss regimen based on exercise, behavior modification and caloric restriction. The usual adult dosage is *** administered either once daily or in divided doses.(1) The mechanism of action of phentermine for weight loss is similar to that of other anorectic agents; it decreases appetite and stimulates the central nervous system.(1) It is postulated that ***, and there is a ***.(3), (4) Additionally, increased *** levels may result in a decrease in *** that may result in increased satiety and decreased appetite.
Topiramate, a sulfamate-substituted monosaccharide, is an anticonvulsant agent indicated as adjunctive therapy for partial onset seizures, primary generalized tonic-clonic seizures, seizures associated with Lennox-Gastaut syndrome and for migraine headache prophylaxis.(5) The
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
recommended total daily dose of topiramate for treatment of seizures in adults is *** administered in two divided doses; ***. Topiramate is known to ***.(5) Recent clinical studies have shown that topiramate can promote weight loss in ***.(16),(17),(18) However, the exact mechanism by which topiramate exerts its anorectic effect is unknown although it is postulated to ***.
A detailed summary of the clinical and nonclinical toxicology, pharmacokinetics and metabolism of phentermine and topiramate is provided in the Investigator Brochure.
VI-0521 has been studied in a Phase 2, randomized, double-blind clinical trial of 200 otherwise healthy obese adults under an Investigator-Initiated investigational new drug application (IND).(19) In this study, subjects were randomized into 1 of 4 treatment groups: ***. Treatment was continued for *** (including ***). Weight loss (percent of total body weight) among subjects treated with VI-0521 ***, compared to *** among *** among ***, and *** among ***. Subjects treated with VI-0521 *** than subjects in the other treatment groups. Significant decreases vs *** and *** were observed in subjects receiving VI-0521. Of the 200 subjects randomized, *** completed treatment to *** group completed treatment. No deaths or serious adverse events were reported during the study and no significant changes in heart valve morphology were observed. The most commonly reported adverse events in subjects treated with VI-0521 were ***.
1.2. Rationale
VI-0521 is an investigational weight loss therapy that is a combination of two currently approved drugs, phentermine and topiramate. As such, VI-0521 represents a potential advance in the medical treatment of obesity since the two agents comprising this combination product affect weight loss through different mechanisms. Additionally, some of the expected side effects of the two drugs may be mitigated by complementary effects of the other. Thus, combining phentermine with topiramate may produce a similar or better adverse event profile compared to either of these agents individually. Topiramate therapy has been associated with ***. Due to the ***, it is mechanistically possible that ***. Thus, the dual mechanisms and low drug doses employed in VI-0521 may provide a safe and effective pharmacotherapy for the achievement and maintenance of weight loss in obese adults.
2. TRIAL OBJECTIVES
The objectives of this study are to evaluate the safety and efficacy of two doses of VI-0521 compared to placebo in the treatment of overweight and obesity in adults with at least *** obesity-related co-morbid conditions.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
3. TRIAL DESIGN
In this prospective, randomized, double-blind, placebo-controlled trial, overweight and obese male and female subjects with at least *** obesity-related co-morbidities who meet the eligibility criteria will be randomly assigned among *** treatment groups shown in Figure 1.
***
Figure 1. Schematic Representation of Study Design.
Approximately 2500 subjects will be randomized *** at up to *** study sites in the United States. The randomization schedule will be stratified by *** to ***; at least *** of subjects will be ***.
Subjects will initiate treatment with a dose titration during weeks 1-4 with doses gradually increased at *** intervals, as determined by randomization assignment, until the specified dose is reached and will be treated for 52 weeks (weeks 5-56) at the assigned dose level. Subjects will return to the site at the end of weeks 2 and 4 during titration and at 4-week intervals thereafter. A schedule of events is provided in Appendix 1.
The primary endpoints are differences between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), and percent of subjects with weight loss of 5% or more at end of treatment (week 56). Percent weight loss will be calculated as ***.
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) for *** and *** groups.
· The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups.
· The difference in change in waist circumference (randomization to week 56) for *** and *** groups.
Additional efficacy endpoints will include the following:
· Effect on ***;
· Effect on *** of *** and ***; ***, *** and ***;
· Change from baseline to week 28 and week 56 in BMI;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· The change in obesity-associated markers (total cholesterol, triglycerides, LDL-C, HDL-C, HgbA1c, fasting glucose, fasting insulin, a measure of insulin sensitivity, systolic blood pressure, diastolic blood pressure, C-reactive protein);
· Change in *** from screening to *** and ***;
· Change from baseline in medication number and dosages for medications to treat cardiovascular or metabolic co-morbidities;
· Difference between *** and *** groups in the rate of progression to type 2 diabetes (subjects non-diabetic at screening); and
· Baseline adjusted change in Framingham 10-year risk score at weeks 28 and 56;
· Change from *** to *** and *** in body composition [assessed by *** at ***].
The change in weight loss (absolute, percent, percent of subjects achieving weight loss of > 5% and > 10% of starting weight), waist circumference and obesity associated risk factors will also be assessed over time. Subgroup analyses, including but not limited to analysis by gender, age and race, may be performed.
Safety evaluations will include assessment of adverse events, including eye symptoms, ***.
*** will also be obtained, and effects of various cofactors including (but not limited to) ***, gender, race, ***, and age will be evaluated. ***.
4. SUBJECT SELECTION
This study is designed to assess the effect of treatment with VI-0521 on overweight and obese adult subjects with *** or more obesity-related co-morbid conditions either currently uncontrolled or requiring measures beyond first line drug treatment to control.
The following eligibility criteria are designed to select subjects for whom protocol treatment is considered appropriate. All relevant medical and non-medical conditions should be taken into consideration when deciding whether this protocol is suitable for a particular subject.
4.1. Inclusion Criteria
To be eligible for enrollment into this trial, subjects must meet all of the following criteria. Specifically, subjects must:
1. Be adults 70 years of age or less;
2. Have a BMI > *** and < *** (for diabetic subjects, there is no lower limit on BMI for study inclusion);
3. Have *** or more of the following obesity-related co-morbid conditions:
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· ***:
· ***;
· ***;
· ***.
· ***;
· ***:
· ***;
· ***
· ***.
· ***;
4. If females of child-bearing potential, be using adequate contraception, defined as double barrier methods, stable hormonal contraception plus single barrier method, or tubal ligation. Females are considered to be of child-bearing potential unless they have undergone a hysterectomy or bilateral oophorectomy, are 55 years of age or greater and have experienced spontaneous cessation of menses for at least 1 year, or have a documented follicle stimulating hormone level >40 IU/L;
5. Provide written informed consent;
6. Be willing and able to comply with scheduled study visits, treatment plan, laboratory tests and other study procedures.
4.2. Exclusion Criteria
Subjects presenting with any of the following will not be included in the trial:
1. Known allergy or hypersensitivity to phentermine or topiramate, any prior use of a combination of phentermine and topiramate for weight loss or use of phentermine or topiramate for any indication within the past 3 months;
2. Weight gain or loss of greater than 5 kg, use of a very low-calorie diet, or participation in a formal weight loss program (investigational or otherwise) within the past 3 months (this includes: Weight Watchers and related dietary/lifestyle intervention programs; prepared food programs; prescribed or over-the-counter weight loss medications; dietary supplement or herbal preparations, teas, or tinctures intended for weight loss; or any supervised fast or very low calorie diet);
3. Obesity of a known genetic or endocrine origin;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
4. History of any eating disorders (e.g. bulimia, binge eating disorder) within the past year;
5. History of drug abuse within the past year;
6. History of alcohol abuse (defined as >14 drinks per week) within the past year;
7. Previous bariatric surgery;
8. Smoking cessation within the previous 3 months or plans to quit smoking during study participation;
9. History of glaucoma, history of increased intraocular pressure or any past or present use of medications to treat increased intraocular pressure;
10. Clinically significant thyroid dysfunction as evidenced by signs or symptoms of hypothyroidism, a TSH > 1.5 x ULN, or use of thyroid hormone treatment that has not been stable for at least 3 months;
11. Use of chronic systemic glucocorticoid therapy, or any other steroid hormone therapy that has not been stable for at least 3 months;
12. Any history of bipolar disorder or psychosis, greater than one lifetime episode of major depression, current depression of moderate or greater severity (PHQ-9 score of 10 or more), presence or history of suicidal behavior or ideation with some intent to act on it, or antidepressant use that has not been stable for at least 3 months;
13. Diagnosis of type 1 diabetes or use of any antidiabetic medication other than metformin within the past month;
14. Stroke, myocardial infarction, life-threatening arrhythmia, coronary re-vascularization within the past 6 months;
15. Unstable angina, congestive heart failure (NYHA Class II, III or IV), or known or suspected clinically significant cardiac valvulopathy;
16. Systolic blood pressure >160 mmHg, diastolic blood pressure > 100 mmHg or antihypertensive medication that has not been stable for at least 1 month;
17. Any history of malignancy within 5 years other than surgically excised basal or squamous cell carcinoma of the skin or cervical cancer;
18. Cholelithiasis within the past 6 months;
19. Any history of nephrolithiasis;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
20. Creatinine clearance < 60 mL/minute;
21. For female subjects of childbearing potential, pregnancy, breastfeeding or plans for pregnancy during the study period;
22. Use of any investigational medication or device for any indication within the last month;
23. Evidence of any clinically significant renal, pulmonary, hepatic, psychiatric or other condition by history, physical examination or laboratory studies that, in the opinion of the investigator, would contraindicate the administration of study medications, affect compliance, interfere with study evaluations or confound the interpretation of study results.
4.3. Randomization Criteria
The following additional criteria must be met prior to randomization and dispensing of study medication to subjects:
1. Baseline physical examination, ECG and laboratory findings with no abnormalities that are considered clinically significant by the principal investigator;
2. Laboratory values that are within the ranges specified below:
***
4.4. Life Style Guidelines
Subjects randomized into the study will receive counseling on how to reduce their caloric intake by 500 kcal/day and will be advised to increase their daily water intake. Subjects will be advised to initiate a lifestyle modification program utilizing the LEARN® Program for Weight Management.(20) The LEARN® program is designed as a 16-week program to aid in weight management by providing tools to facilitate lifestyle, exercise, attitude, relationship and nutrition changes. Each subject will be provided with a LEARN® manual and advised to read and implement the material as appropriate to their individual situation between study visits. Site personnel will discuss these materials with subjects at their regularly scheduled visits. However, no data will be collected to document the level of compliance with the program’s dietary, lifestyle and/or exercise recommendations.
To facilitate discussions between study subjects and research staff, subjects will complete a 24-hour dietary recall at their randomization visit (Visit 2); however, information provided as part of this exercise will be used only for discussions related to recommended dietary modification and will not be retained as documentation. No data will be collected during the study to document the level of compliance with dietary recommendations.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
5. TRIAL TREATMENTS
5.1. Allocation to Treatment
Subjects meeting the randomization requirements specified in Section 4.3 will be randomized to study treatment at visit 2 using a centralized computer-generated randomization system. Eligible subjects will be randomly assigned to *** or *** at a ratio of ***. Randomization will be stratified by ***, and at least *** of subjects will be ***. Both the subject and the study site will be blinded as to subject randomization.
To implement subject randomization among treatment groups, each participating site will be pre-stocked with titration kits corresponding to each treatment group. When a subject qualifies for randomization, site personnel will contact an Interactive Voice Response System (IVRS) and provide the information required regarding the study subject. The randomization assignment will then be made, and the site will be instructed to dispense a specific kit number to the study subject. Additional kits will be shipped to the site to replace those dispensed according to the randomization schedule. When subjects return for subsequent study visits, the IVRS system will also be used to dispense appropriate treatment kits.
5.2. Breaking the Blind
Study medication must not be unblinded during the study unless it is considered absolutely necessary by the investigator for the management of an adverse event or other medical emergency. Under such conditions, the identity of the study treatment will be obtained by contacting the IVRS. Any subject whose treatment assignment has been unblinded must discontinue participation in the study.
VIVUS will be notified of any unblinding of subject treatment group ***. Additionally, investigators are required to ensure that any potential serious adverse events are reported according to the requirements outlined in Sections 8.2 and 8.5 and to provide a written report on the reason for unblinding within ***.
5.3. Drug Supplies
5.3.1. Formulation and Packaging
Medications for this trial will consist of ***. Doses specified for each treatment group will be achieved by varying the *** added to each capsule. Regardless of the dosage assignment, all study treatments will be administered as a ***.
Clinical supplies will be manufactured for VIVUS, Inc. by *** in accordance with current Good Manufacturing Practices. All clinical supplies will be labeled with information required by national regulations. Study drug will be packaged into 2 types of kits; titration kits for use during the first 4 weeks of study therapy when doses are being increased gradually to the final
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
assigned dose, and treatment kits, for use once subjects have been titrated to their assigned dosage of medication. Each titration kit will contain *** for use during weeks 1 through 4 of titration, with each ***. Each *** on the *** will be labeled with the *** and will contain capsules with the dose specified for that week of treatment, as outlined in Table 1.
Table 1: VI-0521 Dosage Strengths by Titration Week for Each Treatment Group
|
|
|
*** |
|
*** |
| ||||||
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
Titration kits will be labeled with the ***. *** will be labeled with the ***.
Each treatment kit will consist of a single bottle *** of study medication at the final treatment dosages shown in Table 1. Each bottle will be labeled with the ***.
5.3.2. Preparation and Dispensing
Clinical supplies provided by the sponsor are to be dispensed only by or under the direct supervision of qualified investigators to subjects meeting the criteria for study entry and in accordance with this protocol. Assignment of specific titration and treatment drug kits to study subjects will require the use of the IVRS; however, no other preparation of clinical supplies is required of the investigational staff.
5.3.3. Administration
Investigators will instruct subjects to take 1 capsule of study medication every morning. When dispensing titration kits, investigators should ensure that subjects understand that each card contains a 4-week supply of medication, and that the capsules must be taken in ***. Investigators will also instruct subjects to return the study medication kit to the site at each site visit. For the week 2 visit, the site will perform drug accountability and redispense *** to the subject for use during titration weeks 3 and 4.
5.3.4. ***
*** is an option for subjects who experience ***. *** is implemented through the ***, and will be done without ***. *** is not an option for subjects who experience ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
When *** is not appropriate or when *** may be required due to events unrelated to subject treatment, subjects may ***. *** are possible with agreement from the medical monitor. All subjects undergoing *** for *** may be *** based on discretion of the PI. If dosing has been ***, a new titration kit should be ordered through *** to ***. Subjects who have a ***. For subjects having a ***. For ***, subjects may ***.
If ***, subjects may be ***. Subjects *** will be encouraged to *** and to continue to make site visits at the regularly scheduled intervals. The last date on which the subject ***. If the subject ***. If the subject elects to withdraw completely from the trial (Section 6.4), the end of treatment (Visit 17) testing should be completed.
5.3.5. Compliance
Subject compliance with study medication will be assessed by ***, and *** should implement any corrective action necessary. Subjects who remain noncompliant with study dosing despite corrective actions by site personnel may be discontinued from the trial.
5.4. Drug Storage and Drug Accountability
All unused study drug must be stored in its packaging at room temperature in a dry, secure area. Access to drug storage areas should be limited to the investigator and designated staff involved with the study. All used and unused drug must be maintained at the study site and made available for audits by VIVUS personnel or their designee.
It should be noted that one component of the study drug combination is a Schedule IV controlled substance. The investigator should take all appropriate measures to control access to and dispensing of study drug.
The investigator must maintain records documenting the amount, condition and date of delivery of all study drug received from the sponsor. In addition, all drug dispensed to study subjects during the course of the study must be ***. Subjects must be instructed to *** by each subject. No investigational drug or packaging, used or unused, may be discarded. All packaging and used and unused drug must be returned to the sponsor upon completion of the study.
5.5. Concomitant Medication(s)
5.5.1. Excluded Medications
Subjects must not take the following medications during their participation in this trial:
· ***;
· ***;
· ***;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· ***;
· ***.
Although subjects requiring antidiabetic medications other than metformin may not be entered into the study, subjects who develop the need for other antidiabetic medications during the course of the trial need not be discontinued. Refer to Section 5.6 for treatment and monitoring recommendations.
5.5.2. Other Restricted Medications
Subjects using *** or allowed *** must be on doses that have been stable for at least 3 months prior to screening. Subjects who develop symptoms indicative of *** during the course of the study need not withdraw but should be evaluated and managed as indicated (Section 5.8).
*** and *** are permitted, provided that the dosage has been stable for at least 1 month prior to screening, and the frequency of use does not exceed twice a week.
All other medications used for the treatment of *** associated with *** must be stable for at least 1 month prior to screening. However, adjustment of these medications is permitted during the trial if the subject’s requirements for treatment change.
5.5.3. Documentation of Concomitant Medication Use
All concomitant medications, including over-the-counter products, vitamins and nutritional/herbal supplements, must be listed on the appropriate case report form (CRF) at trial entry. Any changes in concomitant medication during the course of the trial must be noted on the appropriate CRF.
5.6. Treatment of Diabetes
Subjects who are diabetic at study entry or who become diabetic during the course of the study will be provided with ***. Subjects will be instructed to ***. Diabetic subjects will ***.
*** is suggested as the initial therapy for *** type 2 diabetes ***. ***, including ***, should be reserved for subjects who cannot achieve adequate control with other modes of treatment. *** are prohibited, and subjects requiring treatment with these medications must be discontinued from the trial.
Subjects with consistently elevated fasting blood glucose values should ***. For this study, actionable levels for glucose values are ***. Subjects who note *** fasting glucose values *** in daily glucose monitoring logs during the week prior to a study visit would be considered appropriate for ***. Subjects whose fasting blood glucose ***, or whose blood glucose cannot be adequately controlled with the concomitant treatments allowed in this trial should be
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
discontinued from study treatment and referred back to their primary healthcare provider for additional glycemic management. Subjects may continue attending study visits ***.
During treatment, subjects whose fasting blood glucose is ***.
When discontinuing medications, *** should be discontinued first, followed by ***, and finally ***.
5.7. Treatment of ***
For subjects whose ***. If these medications are already present, ***.
Subjects whose ***, should be discontinued from study treatment and referred back to their primary healthcare provider for more intensive management. Subjects may continue attending study visits ***.
For subjects whose ***. For this trial, it is recommended that *** be the first medications to be reduced or withdrawn followed by ***, and lastly, ***.
5.8. Treatment of ***
Subjects who *** need not be discontinued from therapy. These subjects should be assessed clinically and with laboratory testing ***. Following evaluation, subjects found to be *** may be ***, as appropriate.
6. TRIAL PROCEDURES
This trial is being conducted in conjunction with a companion study (OB-302: A Phase III Randomized, Double-Blind, Placebo-Controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in an Adult Population with BMI > ***) that is of similar design. The distinguishing factors between the two trials are related to the subject populations, specifically, the subjects enrolled into this trial will have a *** associated with obesity than the subjects enrolled into study OB-302. Because there is little overlap between the populations for these two trials, the majority of sites participating in this trial will also be conducting study OB-302. Because results of the screening evaluations may be required to determine which trial a particular subject qualifies for, the initial screening procedures for both of these trials are identical, and will be done under a generic screening informed consent. Once the determination of trial placement has been made a specific informed consent document for that study will be obtained and specific trial evaluations will continue. A schedule of study activities by visit is presented in Appendix 1. A detailed list of these activities is provided in the following sections.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
6.1. Screening
Screening activities will take place at Visit 1 (generic) and Visit 1a (additional screening activities specific for this study). An oral glucose tolerance test (OGTT) will be performed at Visit 1a for all subjects eligible for study participation based on results from Visit 1 screening studies. The screening OGTT test should not be performed until initial screening results have been reviewed.
The screening physical examination and screening ECG may be performed at either Visit 1a or Visit 2 (prior to final evaluation and randomization). These studies should not be performed until initial screening results from Visit 1 have been reviewed.
For purposes of this study, Visit 1a is considered to be a part of Visit 1. In consequence, it is not necessary to obtain information on subject weight, waist circumference, ▇▇▇▇▇ ▇▇▇▇▇, pregnancy test, ***, concomitant medications or adverse events at Visit 1a.
6.1.1. Visit 1
Activities at screening (Visit 1) are:
· Obtain written informed consent for screening evaluations;
· Obtain demographics (including age, race, gender, ethnicity, education, history of smoking, hypertension and dyslipidemia and family history of hypertension, smoking and premature cardiovascular disease) and medical history;
· Assess weight (kg), height (cm), waist circumference (cm) and BMI (note: the BMI calculation must be obtained from the IVRS following informed consent and should be obtained prior to initiating the testing described below);
· Assess ▇▇▇▇▇ ▇▇▇▇▇;
· Record baseline concomitant medications;
· Administer *** and ***;
· Administer ***;
· Assess Inclusion/Exclusion Criteria;
· Obtain blood and urine samples for laboratory testing;
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
· Schedule Visit 1a for OGTT testing in 1 week (± 2 days).
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
6.1.2. Visit 1a
· Obtain study-specific written informed consent;
· Administer ***;
· Perform OGTT for all subjects eligible for participation by results of Visit 1 screening tests;
· Obtain blood and urine samples for biomarkers testing [C-reactive protein, microalbumin (urine) and creatinine (urine)];
· Collect blood sample for HgbA1c and C-peptide;
· Schedule the randomization visit (Visit 2) in 1 week (± 2 days).
6.2. Trial Period
6.2.1. Randomization (Visit 2)
Subjects eligible for treatment will be randomized and study drug dispensed at Visit 2. Activities at randomization are:
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
· Perform complete physical examination (including neurological examination and auscultation for heart sounds);
· Obtain written informed consent for *** procedures and perform *** scan ***. *** scans may be performed between Visits 1 and 2, after results from Visit 1 indicate subject may be eligible to participate;
· Perform 12-lead electrocardiogram;
· Assess adverse events (including eye symptoms), if any;
· Obtain ▇▇▇▇▇ ▇▇▇▇▇;
· Obtain weight and waist circumference measurements;
· Assess changes in concomitant medications;
· Evaluate randomization criteria (Section 4.3);
· Obtain 5 mL serum sample to be retained for possible use for scientific exploratory testing;
· Contact IVRS and obtain subject study medication identification;
· Administer *** for *** and ***;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Complete 24 hour dietary recall, distribute LEARN® materials and perform lifestyle counseling;
· Dispense study medication *** from the assigned kit and provide instructions for proper use;
· Schedule the next study visit in 2 weeks (± 2 days).
6.2.2. Titration (Visit 3)
Subjects will visit the study site at the end of the second week of titration (Visit 3). Activities at Visit 3 are:
· Obtain weight and waist circumference measurements;
· Obtain ▇▇▇▇▇ ▇▇▇▇▇;
· Assess adverse events (including eye symptoms), if any;
· Assess concomitant medications;
· Administer *** and ***;
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
· Review the study medication used since Visit 2; assess treatment compliance and perform drug accountability. Re-dispense the titration card to the subject for use during weeks 3 and 4;
· Perform brief review of lifestyle counseling and answer questions, if any;
· Schedule the next study visit in 2 weeks (± 2 days).
6.2.3. Treatment (Visits 4 through 16)
Subject titration will be completed on Visit 4; subsequently, subjects will return to the site at 4-week intervals for evaluation and to obtain additional study medication. Activities will be as follows:
· Obtain weight and waist circumference measurements;
· Obtain ▇▇▇▇▇ ▇▇▇▇▇;
· Assess adverse events (including eye symptoms), if any;
· Assess concomitant medications;
· Administer *** and ***;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Perform OGTT (Visit 4 only);
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
· Perform brief review of lifestyle counseling and answer questions, if any;
· Obtain fasting blood samples for blood chemistry testing (Visits 4, 5, 7, 10 and 13 only).
· Ensure that the previous dose of trial medications was taken between 20 and 28 hours ago, and obtain a *** blood sample for *** evaluations (if the previous dose of trial medications is outside this window, *** should be postponed for 1 day) (Visits 7 and 10 only);
· Obtain samples for biomarkers: C-reactive protein (blood), microalbumin (urine) and creatinine (urine) (Visit 10 only);
· Obtain samples for HgbA1c for diabetic subjects only (Visits 7 and 13 only);
· Obtain samples for HgbA1c for all subjects (Visit 10 only);
· Obtain blood samples for hematology testing (Visit 10 only);
· Administer *** for *** and *** (Visits 4, 7 and 10 only);
· Administer *** and *** (Visit 10 only);
· Perform *** scan *** (Visit 10 only);
· Collect study medication from previous visit; assess for treatment compliance and perform drug accountability;
· Dispense study medication for the upcoming study interval and provide the proper instructions for use;
· Schedule the next study visit in 4 weeks (± 1 week).
6.2.4. End of Treatment (Visit 17 or Withdrawal from Study)
The end of treatment for subjects completing the study is Visit 17. End of treatment testing will also be performed for subjects who are withdrawn from treatment prior to completion of the study at the time of their treatment termination. For subjects who withdraw from the study prior to completion, the site will also attempt to contact the subject at or about the 56-week time point to obtain a weight measurement, waist circumference, blood chemistry panel and ▇▇▇▇▇ ▇▇▇▇▇.
Activities at the end of treatment visit include:
· Obtain weight and waist circumference measurements;
· Obtain ▇▇▇▇▇ ▇▇▇▇▇;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Assess adverse events (including eye symptoms), if any;
· Assess concomitant medications;
· Administer ***;
· Collect urine sample for pregnancy test (females of childbearing potential only) and perform pregnancy test;
· Administer ***;
· Administer *** and ***;
· Perform *** scan ***;
· Administer *** for *** and ***;
· Complete End of Treatment Questions: ***;
· Obtain fasting blood and urine samples for laboratory testing;
· Obtain samples for biomarkers: C-reactive protein (blood), HgbA1c (blood), microalbumin (urine) and creatinine (urine);
· Perform complete physical examination (including neurological examination and auscultation for heart sounds);
· Perform 12-lead electrocardiogram;
· Collect study medications dispensed at previous visit, assess treatment compliance and perform drug accountability;
6.3. Study Period
The study period for each subject will begin when written informed consent is provided and will continue until Visit 17 (week 56 or early termination) is completed. Sites should link the scheduling of visits to the randomization visit (Visit 2, week 0). Visit windows are provided to allow subject and site scheduling convenience. However, every effort should be made to ensure that visits occur within these windows so that the overall treatment duration is 56 weeks, including titration period, for subjects who complete all visits. In certain instances, adverse event information may be required for events occurring after the study period (Section 8.4).
6.4. Subject Withdrawal
Subjects may withdraw from the trial at any time and for any reason. Additionally, the subject may be withdrawn because of:
· Adverse event;
· Subject lost to follow-up;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Requirement for other medical treatment excluded by the protocol;
· Lack of compliance with the provisions of the protocol;
· Treatment unblinded by investigator;
· Pregnancy;
· Lack of efficacy;
· Termination of trial.
Withdrawn subjects will not be replaced.
Subjects discontinued from treatment should be encouraged to remain in the trial off-treatment and to continue site visits at the scheduled intervals (Section 5.3.3). Subjects who withdraw completely from the trial at any point should complete the end of treatment (Visit 17, week 56) testing. The date of last dose should be recorded.
Every effort should be made to document subject outcome. For subjects who elect to withdraw from the trial without continuing site visits, the investigator should ***.
At about the 56 week time point, withdrawn subjects who have not continued site visits should be asked to return to the site to obtain weight and waist circumference measurements at a minimum and, if possible, the following additional information: adverse events, ▇▇▇▇▇ ▇▇▇▇▇, laboratory tests (chemistry, hematology, urinalysis), concomitant medications, questionnaires ***.
If a subject withdraws from the trial and also withdraws consent for disclosure of future information, ***.
Investigators must discontinue trial participation for all subjects if the sponsor terminates the trial, and evaluations that would normally be performed upon trial completion should be made at that time. For subjects who have received ***, this includes obtaining information requested for the Visit 17 (week 56) end of treatment visit.
7. ASSESSMENTS
7.1. Weight Assessment and Waist Measurement
Subjects will be weighed (kg) and a waist circumference (cm) measurement obtained at each study visit (Visits 1-17). Height (cm) and BMI will be obtained at the screening visit (Visit 1) only.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
7.1.1. Weight Assessment
Subjects should be weighed in kilograms using a calibrated digital scale. The same scale should be used for each measurement and measurements should be evaluated by the same site personnel at each visit, whenever possible. Subject weights should be obtained, whenever possible, under the same conditions (no shoes, clothing of similar weight) that were employed at the first (screening) weighing. Subjects should be encouraged to complete their weigh-in visits in the morning and should be fasting prior to weigh-in.
7.1.2. Waist Measurement
Waist circumference measurements (cm) will be performed using a measuring tape provided by VIVUS, Inc and should be obtained by the same individual at each visit, when possible. To measure the waist circumference, locate the top of the right iliac crest. Place the measuring tape in a horizontal plane (parallel to the floor) around the abdomen at the level of the top of the iliac crest as shown in Figure 2.
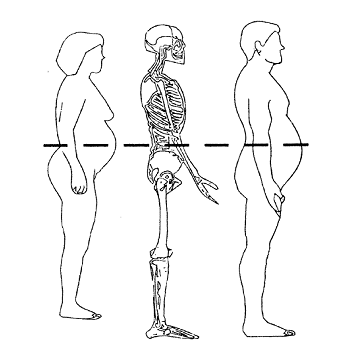
Figure 2. Measuring Tape Position for Waist Circumference Assessments
Ensure that the subject is relaxed. Ensure that the tape is snug but does not indent or compress the skin, and make the measurement (in centimeters) at the end of a normal expiration.(21)
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
7.1.3. Height and BMI
Height measurements (cm) and BMI will be determined by the site at screening only. Height measurements should be made without shoes.
BMI will be calculated for purposes of screening using the IVRS. A printed copy of the BMI obtained from the IVRS should be retained as part of the subject’s source documentation. Manual BMI calculations should not be performed.
If a subject does not meet the BMI criterion for inclusion into the study, no further screening procedures should be undertaken.
7.2. ▇▇▇▇▇ ▇▇▇▇▇
▇▇▇▇▇ ▇▇▇▇▇ (systolic blood pressure, diastolic blood pressure, heart rate, respiration rate, temperature) will be assessed at screening and each study visit. Subjects should be seated comfortably for at least *** prior to assessing ▇▇▇▇▇ ▇▇▇▇▇. Pulse rate and respiratory rate measurements should be made by counting events (heartbeats or breaths) for a period of 30 seconds and multiplying these values by 2 to obtain the rates per minute. A calibrated cuff should be employed for blood pressure measurements. Whenever possible, the same person should perform all assessments for a given subject.
7.3. Questionnaires
7.3.1. ***
The *** for the assessment of ***.(22)-(24)
Because this instrument is intended to be completed ***, it is important that *** on this questionnaire in any way. Should subjects ***. Because this questionnaire assesses the ***. Site personnel, therefore, must carefully review questionnaires for completeness before ***, and assure that questionnaires are properly completed.
The *** is being used ***. This questionnaire will be completed at ***, and at ***after the ***. Answers to the questionnaire may reveal evidence of ***, including the ***. It is the responsibility of the investigator to evaluate ***. The evaluation by the Investigator will be guided by the ***. Investigators should document any such problems ***. It is expected that any randomized ***.
7.3.2. ***
The ***(25) is an ***. Each of the ***, and is answered on a yes/no basis. This assessment will be administered to all *** at *** in order to confirm the *** included in the treatment program. Subsequent *** evaluations will be done at *** after ***. All *** assessments must be
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
administered by a trained interviewer. If any assessments reveal ***, then the results must be reviewed by a physician investigator prior to ***.
7.3.3. ***
The *** that will be completed at *** and ***.
This questionnaire is designed to evaluate the ***.(26), (27) This instrument is intended to be completed ***.
Site personnel must ***. It is critical, therefore, that site personnel review questionnaires for completeness at the time they are initially filled out, and that any missing answers are completed before the ***.
7.3.4. ***
The *** is a *** designed to evaluate ***.(28) This questionnaire is to be completed ***.
As with other questionnaires, site personnel ***.
Site personnel must also ***. It is critical, therefore, that site personnel review questionnaires for completeness at the time they are initially filled out, and that any missing answers are completed before the ***.
7.4. *** and End of Treatment Questions
7.4.1. ***
*** for *** and *** will be assessed at *** and at the ***. For the *** assessment, subjects will *** For the *** assessment, a ***
7.4.2. End of Treatment Questions
At the end of treatment, subjects will be asked to respond to *** questions assessing ***. These questions are:
· ***.
· ***.
· ***
7.5. Laboratory Tests
Laboratory tests will be performed at a licensed, certified central testing laboratory identified by the sponsor. Urine pregnancy tests will be performed at the site using kits supplied by VIVUS,
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Inc. Testing will be conducted according to the schedule provided below. For blood chemistry tests, the subject must have fasted for a minimum of 8 hours before the sample can be drawn.
Testing for phentermine and topiramate will be conducted in a sponsor-approved central laboratory using validated laboratory assays.
7.5.1. Blood Chemistry
Fasting blood chemistries will be evaluated at screening (Visit 1), Visit 4 (week 4), Visit 5 (week 8), Visit 7 (week 16), Visit 10 (week 28), Visit 13 (week 40) and end of treatment (Visit 17 or study withdrawal). Tests will be made for the following: albumin, alkaline phosphatase, alanine transaminase (ALT), aspartate amino transferase (AST), blood urea nitrogen (BUN), serum calcium, serum chloride, serum sodium, bicarbonate, creatinine, creatinine clearance, direct bilirubin, gamma-glutamyl transferase (GGT), glucose, lactate dehydrogenase (LDH), serum phosphorus, serum potassium, total bilirubin, total cholesterol, total protein, uric acid, triglycerides, HDL cholesterol (HDL-C) and LDL cholesterol (LDL-C).
7.5.2. Oral Glucose Tolerance Test
An oral glucose tolerance test (OGTT) will be obtained at Visit 1a (after results from Visit 1 indicate the subject may be eligible for participation), at Visit 4 (week 4) and at end of treatment (Visit 17 or study withdrawal) for all subjects.
The OGTT will use a 75 g glucose loading dose; samples will be obtained at baseline and at 2 hours post dose for evaluation of both glucose and insulin levels.
7.5.3. Hematology
Hematology studies will be evaluated at screening (Visit 1), Visit 10 (week 28) and end of treatment (Visit 17 at week 56 or study withdrawal) for hemoglobin, hematocrit, red blood cell count (RBC), total white blood cell count (WBC), WBC differential (neutrophils, lymphocytes, monocytes, eosinophils, basophils) and platelet count.
7.5.4. Urinalysis
A routine midstream urinalysis with reflex microscopic evaluation will be obtained at screening (Visit 1) and end of treatment (Visit 17 or study withdrawal).
7.5.5. Biomarkers
Appropriate blood and urine biomarkers [C-reactive protein, microalbumin (urine) and creatinine (urine)] will be evaluated at screening (Visit 1a), Visit 10 (week 28) and end of treatment (Visit 17 or study withdrawal). Additionally, adiponectin, intercellular cell adhesion molecule (I-CAM),
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
vascular cell adhesion molecule (V-CAM), retinal binding protein 4 (RBP-4), plasminogen activator inhibitor 1 (PAI-1) and fibrinogen will be evaluated at screening (Visit 1a) and end of treatment (Visit 17 or study withdrawal). With the exception of urine microalbumin and creatinine, all post-screening biomarker results will be blinded to investigators and sponsor.
7.5.6. ▇▇▇▇▇▇▇▇▇▇ ▇▇▇
▇▇▇▇▇▇▇▇▇▇ ▇▇▇ (▇▇▇▇▇▇) testing will be performed at screening (Visit 1a) Visit 10 (week 28) and end of treatment (Visit 17 or study withdrawal) for all subjects. Additionally, HgbA1c testing will be performed at Visit 7 (week 16) and Visit 13 (week 40) for diabetic subjects.
7.5.7. Urine Pregnancy Test
A urine pregnancy test will be obtained for females of childbearing potential at each study visit (Visits 1-17). Urine pregnancy testing will be performed at the site and results reported on the appropriate CRF.
Female subjects who have undergone a hysterectomy or bilateral oophorectomy, who have a follicle stimulating hormone level > 40 IU/L or who are 55 years of age or greater and have experienced cessation of menses for at least 1 year are considered to not be of childbearing potential. Urine pregnancy testing is not required in these subjects.
Management of any subject who becomes pregnant during this study is described in Section 8.5.3.
7.5.8. Thyroid Stimulating Hormone (TSH)
A test for thyroid stimulating hormone (TSH) will be conducted at screening (Visit 1).
7.5.9. Antibody to HIV, HCV, HBsAg
Testing for hepatitis B surface antigen (HBsAg) and antibodies to human immunodeficiency virus (HIV) and hepatitis C virus (HCV) will be conducted at screening.
7.5.10. C-Peptide
A test for C-peptide will be conducted at screening (Visit 1a).
7.5.11. Screening Serum Sample (Retain)
A 5.0 mL serum sample will be obtained at Visit 2 to be retained at the central laboratory for possible use in scientific experimental studies.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
7.5.12. Urine Drug Screen
A urine drug screen will be conducted at screening. Subjects will be tested for cannabinoids, cocaine, amphetamines, opiates and phencyclidine.
7.6. *** Evaluation
Samples for the evaluation of *** will be obtained at ***.
Subjects should be advised at the previous visit to refrain from dosing with study medication until after the clinic visit on *** and should be reminded about 1 week prior to the visit to delay dosing on the days of *** until after testing at the site. If the subject has taken study medication < 20 hours prior to the clinic visit, *** should be postponed until the next day.
7.7. Physical Examination
A complete physical examination will be performed at Visit 2 (prior to review for randomization) and at Visit 17 (week 56) or early withdrawal. The screening examination should not be performed until results of testing at Visit 1 have been reviewed to confirm that the subject may be eligible for treatment.
The physical examination will consist of the following: general appearance, skin, head, eyes, ears, nose, throat, neck (including thyroid), lymph nodes, chest, heart (including auscultation for heart sounds and murmurs), abdomen, extremities, and neurological examination. Subjects will be evaluated for clinically significant abnormalities that would prevent entry into the study and for clinically significant differences between screening and end of treatment.
7.8. Electrocardiogram (ECG)
Twelve-lead electrocardiographic studies will be obtained at Visit 2 (prior to review for randomization) and the end of treatment (Visit 17 or early withdrawal). Screening ECG studies should not be performed until results of testing at Visit 1 have been reviewed to confirm initial subject eligibility for treatment. Whenever possible, ECGs should be obtained in the morning with the timing of the studies matched as closely as possible. Studies will be evaluated for clinically significant abnormalities that would prevent entry into the study and for clinically relevant changes between screening and end of treatment. Parameters including R-R, QRS, QT, and QTc intervals will also be recorded.
7.9. Framingham Risk Score
The Framingham risk assessment evaluates the 10-year risk for development of coronary heart disease.(29) In order to calculate the Framingham risk, the following information will be recorded at screening: demographics (age, gender, race, and ethnicity), medical history (including histories of smoking, diabetes, congestive heart failure, myocardial infarction, hypertension, and
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
dyslipidemia) and family history of premature cardiovascular disease. The Framingham risk assessment will be calculated by VIVUS, Inc. or its designee based on data provided on the CRF.
7.10. Body Composition
At a selected subset of study sites, body composition assessments will be made at baseline (Visit 2), week 28 (Visit 10) and week 56 (Visit 17) using ***. Scans will be collected at a sufficient number of study sites to provide body composition data on approximately *** subjects from this study combined with protocol OB-302. Equipment and procedures used to obtain *** data will be standardized as described in a separate document. All sites will be trained on these procedures prior to performing scans on study subjects. Scans will be read at a central facility and the reader will be blinded.
8. Adverse Event Reporting
8.1. Adverse Events
Adverse events (AEs) are defined as any untoward medical occurrences in subjects administered the trial treatment, whether or not they have a causal relationship to the treatment. All observed or volunteered adverse events regardless of suspected causal relationship to the investigational product must be reported as described in the following sections.
The investigator must pursue and obtain information adequate to describe adverse events, their severity and relationship to study treatment, and their outcomes. Descriptions of neurological or psychological adverse events should be consistent with standard diagnostic criteria and terminology (such as DSM-IV) rather than general reports of symptoms. For adverse events with a causal relationship to the investigational product, follow-up by the investigator is required until the events or their sequelae resolve or stabilize at a level acceptable to the investigator, and VIVUS concurs with that assessment. Investigators must also assess whether adverse events meet the criteria for classification as serious adverse events (see Section 8.2) requiring immediate notification to VIVUS or its designated representative.
8.1.1. Severity Assessment
The investigator will assess the severity of all adverse events using the ***, ***, or *** to describe the maximum intensity of each adverse event. For purposes of consistency, these intensity grades are defined as follows:
· ***;
· ***;
· ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Note the distinction between the severity and the seriousness of an adverse event. A *** is not necessarily a ***. For example, a headache may be *** but would not be classified as serious unless it met one of the criteria for ***.
8.1.2. Causality Assessment
Trial investigators are required to provide an assessment of causality for all adverse events (serious and non-serious) observed during this trial. This assessment will provide a determination of whether, in the investigator’s judgment, there exists a reasonable possibility that the investigational product caused or contributed to an adverse event. For this assessment, investigators must categorize the causality as either “related” or “not related.” For an adverse event to be considered “related” to the trial treatment, there should be evidence that the event follows a reasonable temporal sequence from the administration of trial treatment or that the event follows a known response pattern to the drug. Causality would be further confirmed by improvement in the adverse event upon stopping the trial treatment and reappearance of the event upon rechallenge.
8.1.3. Abnormal Test Findings
The criteria for determining whether an abnormal objective test finding should be reported as an adverse event are as follows:
· Test result is associated with accompanying symptoms, and/or
· Test result requires additional diagnostic testing or medical/surgical intervention, and/or
· Test result leads to a change in trial dosing or discontinuation from the trial, significant additional concomitant drug treatment, or other therapy, and/or
· Test result is considered by the investigator or sponsor to represent a clinically significant finding.
Merely repeating an abnormal test, in the absence of any of the above conditions, does not constitute an adverse event. Any abnormal test result that is determined to be an error does not require reporting as an adverse event.
8.2. Serious Adverse Events
As defined in the Code of Federal Regulations (21 CFR 312.32), a serious adverse event or serious adverse drug reaction is any untoward medical occurrence at any dose that:
· Results in death;
· Is life-threatening (immediate risk of death);
· Requires inpatient hospitalization or prolongation of existing hospitalization;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Results in persistent or significant disability/incapacity;
· Results in congenital anomaly/birth defect.
Important medical events that may not result in death, be life-threatening, or require hospitalization may be considered serious adverse drug experiences when, based on appropriate medical judgment, they may jeopardize the subject and may require medical or surgical intervention to prevent one of the outcomes listed above. Adverse events that, in the investigator’s judgment, significantly jeopardize trial subjects or require medical or surgical intervention in order to prevent any of the outcomes listed above should therefore be reported as serious adverse events.
8.2.1. Definition of Hospitalization
Adverse events reported from clinical trials that result in hospitalization or prolong an existing hospitalization are considered serious. Any initial admission (even if less than 24 hours) to a healthcare facility meets these criteria.
Outpatient ambulatory surgical procedures (same day surgeries) and routine emergency room treatment do not qualify as hospitalizations. Additionally, hospitalization in the absence of a precipitating, clinical adverse event is not in itself a serious adverse event. Examples include, but are not limited to:
· Admission for treatment of a preexisting condition not associated with the development of a new adverse event or with a worsening of the preexisting condition (e.g., for work-up of persistent pre-treatment lab abnormality);
· Administrative admission (e.g., for yearly physical exam);
· Optional admission not associated with a precipitating clinical adverse event (e.g., for elective cosmetic surgery);
· Pre-planned treatments or surgical procedures should be noted in the baseline documentation for the entire protocol and/or for the individual subject.
Diagnostic and therapeutic non-invasive and invasive procedures, such as surgery, should not be reported as adverse events. However, the medical condition for which the procedure was performed should be reported if it meets the definition of an adverse event. For example, an acute appendicitis that begins during the adverse event reporting period should be reported as the adverse event, and the resulting appendectomy should be recorded as treatment of the adverse event.
8.3. Eliciting Adverse Event Information
The investigator is to report all directly observed adverse events and all adverse events spontaneously reported by the trial subject. In addition, trial subjects should be ***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Certain adverse events require prompt and specific action by the investigator in any clinical trial. The following sections describe additional requirements to ensure ***.
8.3.1. Eye Pain
At each visit, subjects will be queried regarding eye symptoms, *** Subject responses will be recorded as adverse events, where appropriate. If any subject reports eye pain and/or ***, the subject should be referred to ***. Treatment with study drug should be discontinued until the ***.
8.3.2. ***
All subjects will be screened for the presence and *** at *** and subsequently at *** after the *** using a validated survey instrument *** designed for assessment of *** in a primary care setting. The *** is a *** module based directly on the diagnostic criteria for ***. *** will also be assessed at *** and at *** following the *** using the ***.
Should this additional assessment indicate the presence of ***. Any such event must be ***. Subjects must be ***.
Any *** must be ***.
8.3.3. Hypoglycemia
Guidelines for the reporting of adverse events based on results obtained in *** maintained by *** are summarized in Table 2.
Table 2. Guidelines for adverse event reports based on ***.
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
8.4. Reporting Period
The reporting period for adverse events begins when the subject provides written informed consent and extends until *** after the last dose of the investigational product is administered. All adverse events that occur during this period and are known to the investigator must be reported according to the requirements outlined in Section 8.5.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
8.5. Reporting Requirements
All adverse events will be reported on the adverse event page(s) of the CRF. In addition, serious adverse events must also be reported on a separate Serious Adverse Event form. Where the same data are collected, the forms must be completed in a consistent manner. For example, the same adverse event term should be used on both forms. Adverse events should be reported using concise medical terminology on the CRFs as well as on the form for collection of serious adverse event information.
8.5.1. Serious Adverse Event Reporting Requirements
If a serious adverse event occurs, VIVUS, Inc. or designee is to be notified within 1 business day of awareness of the event by the investigator. In particular, if the serious adverse event is fatal or life-threatening, notification to VIVUS, Inc. or designee must be made immediately, irrespective of the extent of available adverse event information. This timeframe also applies to additional new information (follow-up) on previously forwarded serious adverse event reports.
***
For all serious adverse events, the investigator is obligated to pursue and provide information to VIVUS, Inc. or designee in accordance with the timeframes for reporting specified above. In addition, an investigator may be requested by VIVUS, Inc. or designee to obtain specific additional follow-up information in an expedited fashion. This information may be more detailed than that captured on the adverse event case report form. In general, this will include a description of the adverse event in sufficient detail to allow for a complete medical assessment of the case and independent determination of possible causality. Information on other possible causes of the event, such as concomitant medications and illnesses, must be provided. In the case of a subject death, a summary of available autopsy findings must be submitted as soon as possible to VIVUS, Inc. or its designated representative.
8.5.2. Non-Serious Adverse Event Reporting Requirements
Non-serious adverse events are to be reported on the adverse event CRFs, which are to be submitted to VIVUS, Inc. or its designee.
8.5.3. ***
If any trial subject becomes or is found to be *** while receiving the investigational product, the investigator must submit this information to VIVUS, Inc. on an ***.
The investigator will follow the *** and then notify VIVUS, Inc. or its designee of the outcome. The investigator will provide this information as a follow up to the ***.
For reported ***. The status of an ***.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
If *** meet the criteria for immediate classification as a serious adverse event ***, the investigator should follow the procedures for reporting serious adverse events. Similarly, any *** that are considered to be adverse events should be reported as such on the appropriate CRF. However, *** need not be reported as an adverse event if there is no associated adverse outcome.
For reporting purposes, *** should be reported as serious adverse events, but because the ***.
9. DATA ANALYSIS/STATISTICAL METHODS
9.1. Sample Size Determination
In a previous *** study evaluating the effects of VI-0521, subjects treated with a *** had a mean (SD) weight loss of *** for subjects receiving *** treated subjects. If similar standard deviations are achieved in the present trial, the planned sample size of at least 500 subjects per treatment group should provide *** power to detect these differences.
9.2. Efficacy Analysis
9.2.1. Analysis of Primary Endpoints
The primary hypotheses are:
***
The primary calculated endpoints for the trial are the percent weight loss at week 56 calculated as *** and the percentage of subjects achieving at least 5% weight loss at week 56.
The intent to treat (ITT) population (Section 9.2.1.3) is the primary analysis population. For subjects who discontinue treatment prior to trial completion, every attempt will be made to have them return to the clinic at week 56 for a final weight assessment, regardless of when they discontinued treatment. For subjects who ***.
Comparisons between treatments for percent weight loss will be assessed using an *** with factors of ***, ***and ***, and with ***. Comparisons between treatments for the percentage of subjects achieving at least 5% weight loss will be assessed by *** with ***, ***, and *** and ***. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant for both co-primary end points at ***, then the test will proceed to the *** also at ***. If the statistical comparison is not significant at the *** for the *** when compared with ***, then the statistical test will be stopped and the *** will not be tested.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
If both dose groups are significantly better than ***, then the *** and *** dose groups will be compared. The *** for the difference in the mean percent body weight reduction between treatment groups will be derived.
The cumulative probability distribution as a function of percentage change in body weight for each treatment group will be plotted.
9.2.2. Analysis of Secondary Endpoints
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) for *** and *** groups.
· The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups.
· The difference in change in waist circumference (randomization to week 56) for *** and *** groups.
The ITT population will be employed to evaluate all secondary efficacy endpoints, and a step down strategy analogous to that used for the primary endpoint will be implemented to protect the overall alpha levels for these.
The difference in absolute weight reduction and the difference in waist reduction at week 56 from baseline will also be compared using the same *** as the primary end point. *** with ***, *** and *** and ***, will be used to compare the probability of reaching 5% and 10% body weight reduction from baseline to week 56 between treatment groups.
9.3. Analysis of Other Endpoints
9.3.1. Other Efficacy Endpoints
The changes in primary and secondary outcomes over monthly intervals during the study will be evaluated. The methodology for these comparisons will be similar to that used for primary and secondary endpoints. Details of these analyses will be provided in the Statistical Analysis Plan.
The *** data will consist of ***, each of which will be analyzed separately, and a ***. Similarly, the *** will consist of ***. These ***. Changes from baseline will be summarized using *** and ***. Effect sizes (change divided by baseline standard deviation) will also be summarized. Changes from baseline will be compared between treatment groups using *** as appropriate for *** and for *** assessments of eating behavior. Results of the *** questions will be calculated for each study group and compared.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Changes in BMI between baseline and Visits 10 and 17 (weeks 28 and 56) and change in Framingham 10-year risk assessments between baseline and Visits 10 and 17 (weeks 28 and 56) will be evaluated. For these analyses, BMI and Framingham risk scores will be calculated by the central analysis group.
The change from baseline to Visit 17 (week 56) in the number and dosage of medications used to treat cardiovascular or metabolic risk factors will be calculated.
In the *** of subjects treated at ***, changes from *** to *** and *** in percent lean body mass and percent *** will be evaluated. Differences between treatment groups will be evaluated using methods similar to those used to evaluate other continuous variables. For these evaluations, data will be pooled from protocols OB-302 and OB-303.
9.3.2. *** Analysis
*** will be obtained during this trial using a multiple trough sampling scheme, where samples are obtained from each subject at ***. These data will be combined for analyses with data from other Phase 3 trials that will utilize similar sampling schemes. *** analyses will characterize the *** for both drugs. The effects of various covariates including (but not limited to) ***, gender, race, *** and age will be evaluated.
9.4. Analysis Populations
Intent-to-treat (ITT) and safety populations: All subjects who are randomized, take one or more doses and have at least one post dosing assessment will be included in the ITT and safety populations.
9.5. ***
For those subjects who ***. If a subject ***.
9.6. ***
***
9.7. Safety Analysis
Safety analyses will be performed on the safety population and will include all data on all doses reported during the study.
9.7.1. Adverse Events
Adverse events will be coded using a MedDRA coding dictionary. The number and percentage of subjects who reported at least one adverse event in each system organ class and preferred term
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
category, and the total number and percentage of subjects with any AE over all system organ classes will be summarized by treatment group.
Subsets of AEs that are considered serious or required discontinuation of the study medication will be listed by subject and presented separately.
9.7.2. Clinical Laboratory Tests
A summary of observed values and change from baseline will be presented for all laboratory parameters with numerical measures using descriptive statistics. Shift tables displaying low-normal-high at baseline versus low-normal-high at end of treatment in a 3-by-3 contingency table will be provided. For selected laboratory parameters, scatter plots of baseline versus week 56 results, will be produced by treatment group.
A laboratory value that is above or below normal range will be considered an abnormal value. For selected laboratory parameters, threshold limits of clinical concern will be defined as multiplicative factors of the normal ranges. The list of multiplicative factors for each laboratory parameter will be included in the Statistical Analysis Plan. The frequency and percentage of subjects with laboratory results above or below the normal range and threshold limits at each scheduled assessment or any time during the treatment will be summarized by treatment group.
9.7.3. ▇▇▇▇▇ ▇▇▇▇▇ and Other Safety Evaluations
Mean blood pressures, heart rate, respiration rate and temperature, obtained at each visit, will be summarized and plotted by treatment group. Medications, other than study medication, taken during the study will be considered as concomitant medications. They will be summarized by treatment group according to the preferred terms, using the World Health Organization (WHO) Drug Dictionary.
9.7.4. Questionnaire Assessments
Changes in total *** will be summarized by treatment group *** and ***. Since the proposed indication for the study medication is weight loss, ***. Descriptive summaries of individual questionnaire items will also be presented.
9.8. Interim Analysis
An interim analysis may be conducted once approximately half of the study subjects have completed the intended course of treatment to provide information for the planning of future clinical trials that may be designed or commenced prior to the completion of this trial. No changes will be made to the conduct of the present study, including premature termination or increased sample size, based on the results of this interim analysis and no adjustments to the significance level for the primary hypothesis will be made if this interim analysis is conducted.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
9.9. ***
***
10. QUALITY CONTROL AND QUALITY ASSURANCE
During trial conduct, VIVUS, Inc. or its agent will conduct periodic monitoring visits to ensure that the protocol and GCPs are being followed. The monitors may review source documents to confirm that the data recorded on CRFs are accurate. The investigator and institution will allow VIVUS, Inc. monitors or its agents and appropriate regulatory authorities direct access to all appropriate source documents to perform this verification.
The trial site and trial-related documents may be subject to review by the institutional review board (IRB)/independent ethics committee (IEC), and/or to quality assurance audits performed by VIVUS, Inc. or its agents and/or to inspection appropriate regulatory authorities. Refer to Section 13.
It is important that the investigator(s) and their relevant personnel are available during the monitoring visits and possible audits or inspections and that sufficient time is devoted to the process by the investigator and site personnel.
11. DATA HANDLING AND RECORD KEEPING
11.1. Case Report Forms / Electronic Data Record
As used in this protocol, the term case report form (CRF) should be understood to refer to either a paper form or an electronic data record or both, depending on the data collection method used in this trial.
A CRF is required and should be completed for each included subject. The completed original CRFs are the sole property of VIVUS, Inc. and should not be made available in any form to third parties, except for authorized representatives of VIVUS, Inc. or appropriate regulatory authorities, without written permission from VIVUS, Inc.
It is the investigator’s responsibility to ensure CRF completion and to review and approve all CRFs. CRFs must be signed by the investigator or by an authorized staff member. These signatures serve to attest that the information contained on the CRFs is complete and accurate. At all times, the investigator has final personal responsibility for the accuracy and authenticity of all clinical and laboratory data entered on the CRFs. Subject source documents are the physician’s subject records maintained at the trial site. In most cases, the source documents will be the hospital’s or the physician’s chart. In cases where the source documents are the hospital or the physician’s chart, the information collected on the CRFs must match those charts.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
11.2. Record Retention
To enable evaluations and/or audits from regulatory authorities or VIVUS, Inc., the investigator will keep records, including the identity of all participating subjects (sufficient information to link records, eg, CRFs and hospital records), all original signed informed consent forms, copies of all CRFs, serious adverse event forms, source documents, and detailed records of treatment disposition. The records should be retained by the investigator according to specifications in the ICH guidelines, local regulations, or as specified in the Clinical Study Agreement, whichever is longer. The investigator must obtain written permission from VIVUS before disposing of any records, even if retention requirements have been met.
If the investigator relocates, retires, or for any reason withdraws from the trial, VIVUS, Inc. should be prospectively notified. The trial records must be transferred to an acceptable designee, such as another investigator, another institution, or to VIVUS, Inc.
12. ETHICS
12.1. Institutional Review Board (IRB)/Independent Ethics Committee (IEC)
Regulations require that an IRB/IEC oversee all investigational drug studies. This board or committee, the makeup of which must conform to local and regional regulations, will approve all aspects of the study, including the protocol, advertising and written informed consent form to be used prior to initiation of the study. It is the responsibility of the investigator to have prospective approval of the trial protocol, protocol amendments, informed consent forms and other relevant documents, eg, advertisements, if applicable, from the IRB/IEC. All correspondence with the IRB/IEC should be retained in the Investigator File. Copies of IRB/IEC approvals should be forwarded to VIVUS, Inc. or designee. All correspondence with the IRB/IEC should be retained in the Investigator file and copies forwarded to VIVUS, Inc. or designee.
All amendments to the protocol must be reviewed and approved by VIVUS, Inc. and the IRB/IEC prior to implementation. The only circumstance in which an amendment may be initiated prior to IRB/IEC approval is where the change is necessary to eliminate apparent immediate hazards to the subjects. In that event, the investigator must notify the IRB/IEC and VIVUS, Inc. in writing within 5 working days after the implementation.
The investigator is responsible for obtaining annual (at a minimum) IRB/IEC renewal for the duration of the study. The investigator is also responsible for keeping the IRB/IEC advised of the progress of the study, of any changes made to the protocol as deemed appropriate, but at least once a year. Copies of the investigator’s report and of the IRB/IEC extension approval must be forwarded to VIVUS, Inc. or designee.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
12.2. Ethical Conduct of the Trial
The trial will be performed in accordance with the protocol, International Conference on Harmonization Good Clinical Practice (ICH-GCP) guidelines and applicable local regulatory requirements and laws.
12.3. Subject Information and Consent
The informed consent form and any changes to the informed consent form made during the course of the trial must be agreed to by VIVUS, Inc. or designee and the IRB/IEC prior to its use and must be in compliance with all ICH-GCP, local regulatory requirements and legal requirements.
The investigator must ensure that each trial subject is fully informed about the nature and objectives of the trial and possible risks associated with participation and must ensure that the subject has been informed of his/her rights to privacy. The investigator will obtain written informed consent from each subject before any trial-specific activity is performed and should document in the source documentation that consent was obtained prior to enrollment in the trial. The original signed copy of the informed consent form must be maintained by the investigator and is subject to inspection by a representative of VIVUS, Inc., their representatives, auditors, the IRB/IEC and/or regulatory agencies. A copy of the signed informed consent form will be given to the subject.
12.4. Disclosure of Data
Data generated by this trial must be available for inspection by the U.S. Food and Drug Administration (FDA), by the sponsor or a designate acting on behalf of the sponsor, by applicable foreign health authorities, and by the IRB or IEC as appropriate. At a subject’s request, medical information may be given to their personal physician or other appropriate medical personnel responsible for their welfare.
Subject medical information obtained during the course of this trial is confidential and disclosure to third parties other than those noted above is prohibited.
13. REGULATORY CORRESPONDENCE
The trial site and trial-related documents may be subject to review by the IRB/IEC and/or to quality assurance audits performed by VIVUS, Inc. or its designee and/or to inspection by the FDA and/or applicable foreign health authorities. The investigator will notify VIVUS, Inc. within *** working days following any FDA or other regulatory agency contact with the investigative site regarding this study. The investigator will provide VIVUS with copies of all correspondence with the FDA or other regulatory agency which may affect the review of the current study (e.g., Form 483, Inspection Observations) or their qualification as an investigator in studies conducted by VIVUS, Inc. (e.g., warning letters).
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
14. Definition of end of trial
The end of trial is defined as the date when the last subject completes the last trial visit.
Additionally, data and materials that are required by the Sponsor before any trial site’s activity can be considered complete include:
· All completed Case Report Forms, appropriately signed by the investigator;
· All laboratory findings, clinical data and special test results collected during the trial period;
· Completed drug accountability and investigational materials return records;
· Statement of outcome for any serious adverse events reported during the study;
· Copy of notification to IRB or IEC indicating study completion.
15. SPONSOR DISCONTINUATION CRITERIA
Premature termination of this clinical trial may occur because of a regulatory authority decision, change in opinion of the IRB/IEC, drug safety problems, or at the discretion of VIVUS, Inc. In addition, VIVUS, Inc. retains the right to discontinue development of VI-0521 at any time.
If a trial is prematurely terminated or discontinued, VIVUS, Inc. will promptly notify the investigator. After notification, the investigator must contact all participating subjects within 15 days. As directed by VIVUS, Inc., all trial materials must be collected and all CRFs completed to the greatest extent possible.
16. PUBLICATION OF TRIAL RESULTS
All information and data, including the terms of this protocol, and all data, clinical results, and research conducted hereunder concerning VIVUS, Inc’s products and operations including VIVUS, Inc. patent applications, formulas, manufacturing processes, basic scientific data, and formulation information that has been supplied by VIVUS, Inc. and not previously published are considered confidential by VIVUS, Inc. and will remain the sole property of VIVUS, Inc. The investigator understands and agrees that said proprietary and/or confidential information disclosed to or produced by him/her thereunder is highly valuable to VIVUS, Inc. and will be used exclusively by the investigator in accomplishing this study and will not be used for any other purposes without VIVUS, Inc’s prior written consent. The investigator agrees that he/she will not use any such proprietary and/or confidential information for any other purpose. The investigator also understands and agrees that such disclosure will not be deemed to grant to the investigator a license for use of said proprietary and/or confidential information, except as expressly provided herein.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
It is understood by the investigator that the information developed in the clinical study will be used by VIVUS, Inc. in connection with the development of this product. This information, therefore, may be disclosed and used solely by VIVUS, Inc. as required to such third parties and agencies as VIVUS, Inc., in its sole discretion, warrants. In order to allow for the use of the information derived from the clinical studies, it is understood that there is an obligation to provide to VIVUS, Inc. complete test results and all data developed in this study. The investigator agrees to promptly answer all inquires from VIVUS, Inc. regarding completion, legibility or accuracy of trial data in the case report.
VIVUS, Inc. recognizes the value of disseminating research results and expects that publication of all results from this study will be undertaken by a collaborative group of study investigators who made significant contributions to the study design, the treatment of study subjects, and evaluation of study data. However, after 1) submission of the multicenter results for publication, 2) notification ***.
Investigators shall furnish the *** with a written copy of any proposed publication or other disclosure of study results (including disclosures at research seminars, lectures and professional meetings) *** prior to submission for publication or disclosure so that *** may have a reasonable opportunity to protect its proprietary rights to information, inventions, or products developed under this study and to insure that reported data are factually correct. Upon the ***, the investigator shall not publish or disclose information related to this study. Further, if the *** believes that such publication or disclosure contains confidential information, the investigator agrees to remove such confidential information from the proposed publication or disclosure.
VIVUS, Inc. agrees that before it publishes any results of this study in a refereed journal, it will provide the investigator, for review, a prepublication manuscript *** prior to the submission of the manuscript to the publisher.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
17. REFERENCES
(1) Adipex-P® (Phentermine Hydrochloride USP, 37.5 mg) Package Insert (2005). Gate Pharmaceuticals, Sellersville, PA.
(2) ▇▇▇▇▇▇ D, ▇▇▇ M. Obesity and Regulation of Body Mass. Lehninger Principles of Biochemistry. 4th ed. New York: ▇▇ ▇▇▇▇▇▇▇ and Company; 2005:912-913.
(3) Montague C, Farooqi I, ▇▇▇▇▇▇▇▇▇ J, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997;387:903-908.
(4) Heymsfield S, ▇▇▇▇▇▇▇▇▇ A, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: A randomized, controlled, dose-escalation trial. JAMA 1999;282:1568-1575.
(5) Topamax® (topiramate) Tablets and Topamax® (topiramate capsules) Sprinkle Capsules Package Insert (June 2003). Ortho-▇▇▇▇▇▇ Pharmaceutical, Inc., ▇▇▇▇▇▇▇, ▇▇ ▇▇▇▇▇.
(6) ▇▇▇▇▇▇ K, ▇▇▇▇▇▇▇ M, ▇▇▇▇▇ C, ▇▇▇▇▇▇▇▇ C. Prevalence and trends in obesity among US adults, 1999-2000. JAMA 2002;288:1723-1727.
(7) ▇▇▇▇▇▇▇ P, ▇▇▇▇▇ T, ▇▇▇▇ G, et al. Obesity and cardiovascular disease: Pathophysiology, evaluation, and effect of weight loss: An update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Commission of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 2006;113:898-918.
(8) ▇▇▇▇▇ C, ▇▇▇▇▇▇▇ M, ▇▇▇▇▇▇ L, et al. Prevalence of overweight and obesity in the ▇▇▇▇▇▇ ▇▇▇▇▇▇, ▇▇▇▇-▇▇▇▇. JAMA 2006;295:1549-1555.
(9) Must A, Spadano J, ▇▇▇▇▇▇▇ E, et al. The disease burden associated with overweight and obesity. JAMA 1999:282;1523-1529.
(10) Katzmarzyk P, ▇▇▇▇▇▇▇ I, Ardern C. Physical inactivity, excess adiposity and premature mortality. Obes Res 2003;4:257-290.
(11) ▇▇▇▇▇ K, ▇▇▇▇▇▇▇▇▇ A, ▇▇▇▇▇ T, et al. Overweight, obesity, and mortality in a large prospective cohort of persons 50 to 71 years old. NEJM 2006;355:763-778.
(12) ▇▇▇▇▇▇▇▇▇ D. Beneficial health effects of modest weight loss. Int J Obes 1992;6:297-415.
(13) Pasanisi F, Contaido F, ▇▇▇▇▇▇▇▇ G, ▇▇▇▇▇▇▇ M. Benefits of sustained moderate weight loss in obesity. Nutr Metab Cardiovasc Dis 2001;11:401-406.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
(14) Douketis J, ▇▇▇▇▇ C, Thabane L, ▇▇▇▇▇▇▇▇▇▇ D. Systematic review of long-term weight loss studies in obese adults: Clinical significance and applicability to clinical practice. Int J Obes 2005;10:1153-1167.
(15) ▇▇▇▇▇▇ R, Majumdar S. Drug treatments for obesity: Orlistat, sibutramine, and rimonabant. Lancet 2007;369:71-77.
(16) ▇▇▇-▇▇▇▇▇▇▇▇ E, ▇▇▇▇▇▇▇ M, ▇▇▇▇▇▇▇▇ E, et al. Predictors of weight loss in adults with topiramate-treated epilepsy. Obes Res 2003;11:556-562.
(17) ▇▇▇▇ G, ▇▇▇▇▇▇▇▇▇ P, ▇▇▇▇▇ S, et al. A 6-month randomized, placebo-controlled, dose-ranging trial of topiramate for weight loss in obesity. Obes Res 2003;11:722-733.
(18) ▇▇▇▇▇▇▇ J, ▇▇▇▇▇▇▇ L, ▇▇▇▇▇▇▇▇ A, et al. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of topiramate in the treatment of obese subjects. Int J Obes Relat Metab Disord 2004;28:1399-1410.
(19) Clinical Study Report: A Phase II, 24-Week, Randomized, Double-Blind, Placebo-Controlled, Single-Center Study to Examine Safety, Tolerability and Efficacy of Topiramate and Phentermine Combination Therapy for Weight Loss in Healthy Obese Subjects. Data on file. VIVUS, Inc., Mountain View, CA.
(20) The LEARN® Program for Weight Management 2000. ▇▇▇▇▇ ▇. ▇▇▇▇▇▇▇▇. The Life Style Company™. Dallas.
(21) National Heart, Lung and Blood Institute Obesity Education Initiative. The Practical Guide: Identification, Evaluation and Treatment of Overweight and Obesity in Adults. NIH Publication No. 00-4084. October 2000.
(22) ***
(23) ***
(24) ***
(25) ***
(26) ***
(27) ***
(28) ▇▇▇▇ ▇▇, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Medical Care 1992; 30:473-83.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
(29) ▇▇▇▇▇▇ P, ▇’▇▇▇▇▇▇▇▇ R, ▇▇▇▇ D, et al. Prediction of Coronary Heart Disease Using Risk Factor Categories. Circulation 1998;97:1837-1847.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 1: SCHEDULE OF STUDY ACTIVITIES
|
|
|
*** |
|
*** |
|
*** |
|
*** |
| ||||||||||||||||||||||||||||
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
*** |
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
|
|
*** |
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** |
|
|
*** |
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
|
|
*** |
|
|
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
|
|
|
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
*** |
|
|
*** |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 2: ***
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 3: ***
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 4: ***
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
APPENDIX 5: PROTOCOL AMENDMENTS
Amendment Tracking
Protocol Title: A Phase III Randomized, Double-Blind, Placebo controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in Adults with Obesity-Related Co-Morbid Conditions
Protocol Number: OB-303
Indicate any amendments to this protocol by writing the Amendment Number and the Date of the Amendment in the space below. This page will be used to track the original protocol and its amendments. This is the original protocol if no amendment number is listed below.
Amendment Number 3
Date of Amendment: ***
(19 items)
Rationale: This protocol amendment is being implemented at the request of the FDA to incorporate assessments of *** and *** using the ***, and assessments of *** using the *** in all study subjects at ***. Previously, follow-up *** evaluations were done only if other *** assessments revealed a ***. This amendment also provides for body composition assessments at selected sites using ***. These changes are not anticipated to have any impact on the safety of study subjects.
|
Section and/or Item |
|
Protocol |
|
Amendment 3, *** |
|
Protocol Synopsis: Efficacy Endpoints — Secondary efficacy endpoints are: |
|
*** |
|
Added: “Effect on body composition as indicated by ***. *** assessments will be performed at only at a ***.” |
|
|
|
|
|
|
|
Protocol Synopsis: Safety Endpoints |
|
*** |
|
Change: “***”
To: “Follow-up assessments will be done at *** after ***.” |
|
|
|
|
|
|
|
List of Abbreviations |
|
*** |
|
Added: “***” |
|
|
|
|
|
|
|
Rationale |
|
*** |
|
Change: “VI-0521 is an exploratory weight loss therapy that is a new combination of two currently approved drugs, phentermine and topiramate.”
To: “VI-0521 is an investigational weight loss therapy that is a combination of two currently approved drugs, phentermine and topiramate.” |
|
|
|
|
|
|
|
Trial Design: Additional |
|
*** |
|
Added: “Change from *** to *** and *** in body composition [assessed by |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
efficacy endpoints will include the following: |
|
|
|
*** at ***].”
Change: “Safety evaluations will include assessment of adverse events, including eye symptoms, ***”
To: “Safety evaluations will include assessment of adverse events, including eye symptoms, ***” |
|
|
|
|
|
|
|
Trial Period: Randomization (Visit 2) |
|
*** |
|
Added: “Obtain written informed consent for *** procedures and perform *** scan ***. *** scans may be performed between Visits 1 and 2, after results from Visit 1 indicate subject may be eligible to participate;”
Change: “Assess adverse events (including eye symptoms and ***), if any;”
To: “Assess adverse events (including eye symptoms), if any;” |
|
|
|
|
|
|
|
Trial Period: Titration (Visit 3) |
|
*** |
|
Added: “Administer *** and ***;”
Change: “Assess adverse events (including eye symptoms and ***), if any;”
To: “Assess adverse events (including eye symptoms), if any;” |
|
|
|
|
|
|
|
Trial Period: Treatment (Visits 4 through 16) |
|
*** |
|
Added: “Administer *** and ***;”
Delete: “Administer *** (Visits 7, 10, and 13 only);”
Change: “Assess adverse events (including eye symptoms and ***), if any;”
To: “Assess adverse events (including eye symptoms), if any;”
Added: “Perform *** scan *** (Visit 10 only);” |
|
|
|
|
|
|
|
End of Treatment (Visit 17 or Withdrawal from Study) |
|
*** |
|
Added: “Administer ***;”
Change: “Assess adverse events (including eye symptoms and ***), if any;”
To: “Assess adverse events (including eye symptoms), if any;”
Added: “Perform *** scan ***;” |
|
|
|
|
|
|
|
Subject Withdrawal |
|
*** |
|
Change: “At about the 56 week time point, withdrawn subjects who have not continued site visits should be asked to return to the site to obtain weight and waist circumference measurements at a minimum and, if possible, the following additional information: adverse events, ▇▇▇▇▇ ▇▇▇▇▇, laboratory tests (chemistry, hematology, urinalysis), concomitant medications, questionnaires ***.”
To: “At about the 56 week time point, withdrawn subjects who have not continued site visits should be asked to return to the site to obtain weight and waist circumference measurements at a minimum and, if possible, the following additional information: adverse events, ▇▇▇▇▇ ▇▇▇▇▇, laboratory tests (chemistry, hematology, urinalysis), concomitant medications, questionnaires ***.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “The *** is being used to ***. This questionnaire will be completed at |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
*** and at ***.”
To: “The *** is being used to ***. This questionnaire will be completed at ***, and at *** after the ***.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “Subsequent *** evaluations will be done only in *** who demonstrate a ***.”
To: “Subsequent *** evaluations will be done at *** after ***. All *** assessments must be administered by a trained interviewer. If any assessments reveal ***, then the results must be reviewed by a physician investigator prior to ***.” |
|
|
|
|
|
|
|
Body Composition |
|
*** |
|
Added New Section:
“At a selected subset of study sites, body composition assessments will be made at baseline (Visit 2), week 28 (Visit 10) and week 56 (Visit 17) using ***. Scans will be collected at a sufficient number of study sites to provide body composition data on approximately *** subjects from this study combined with protocol OB-302. Equipment and procedures used to obtain *** data will be standardized as described in a separate document. All sites will be trained on these procedures prior to performing scans on study subjects. Scans will be read at a central facility and the reader will be blinded to the identity of the subjects, the treatment assignment, and the visit number.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “All subjects will be screened for the presence and *** at *** and subsequently at regular intervals throughout the study: ***, and *** using a validated survey instrument *** designed for assessment of *** in a primary care setting.”
To: “All subjects will be screened for the presence and *** at *** and subsequently at *** after the *** using a validated survey instrument *** designed for assessment of *** in a primary care setting.”
Change: “The *** is a *** module based directly on the diagnostic criteria for ***. At study visits where *** assessments are not included, subjects will be asked the following question to assess ***.”
To: “The *** is a *** module based directly on the diagnostic criteria for ***. *** will also be assessed at *** and at *** following the *** using the ***.”
Change: “Any *** must be ***. Investigators are encouraged to administer the *** on an ad-hoc basis as part of the clinical assessment of ***. These ad-hoc questionnaires will be maintained as source documentation, but will not be analyzed as a separate ***.”
To: “Any *** must be ***.” |
|
|
|
|
|
|
|
Other Efficacy Endpoints |
|
*** |
|
Added: “In the *** of subjects treated at ***, changes from *** to ***and *** in percent lean body mass and percent *** will be evaluated. Differences between treatment groups will be evaluated using methods similar to those used to evaluate other continuous variables. For these |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
evaluations, data will be pooled from protocols OB-302 and OB-303.” |
|
|
|
|
|
|
|
Appendix 1: Schedule of Study Activities |
|
*** |
|
Added: “***” and only marked at *** |
|
|
|
|
|
|
|
Appendix 1: Schedule of Study Activities — *** |
|
*** |
|
Added at *** |
|
|
|
|
|
|
|
Appendix 1: Schedule of Study Activities — *** |
|
*** |
|
Added at *** |
|
|
|
|
|
|
|
Appendix 1: Schedule of Study Activities — *** |
|
*** |
|
Change: “***”
To: “***” |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Protocol Title: A Phase III Randomized, Double-Blind, Placebo controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in Adults with Obesity-Related Co-Morbid Conditions
Protocol Number: OB-303
Indicate any amendments to this protocol by writing the Amendment Number and the Date of the Amendment in the space below. This page will be used to track the original protocol and its amendments. This is the original protocol if no amendment number is listed below.
Amendment Number 2
Date of Amendment: ***
(12 items)
Rationale: This amendment is being implemented to address inconsistencies in the description of various study exclusions and analyses, and to specify that an ***. Various typographical errors in the previous version of the protocol have also been corrected. None of the changes implemented with this amendment are anticipated to have any impact on safety risks to the study subjects.
|
Section and/or Item |
|
Protocol Date |
|
Amendment 2, *** |
|
Protocol Synopsis: Safety Endpoints |
|
*** |
|
Change: “Safety will be assessed by an evaluation of adverse events, including eye symptoms (collected each study visit); ***;”
To: “Safety will be assessed by an evaluation of adverse events, including eye symptoms (collected at each study visit); ***;” |
|
|
|
|
|
|
|
Protocol Synopsis: Statistical Methods |
|
*** |
|
Change: “All subjects who are randomized, take one or more doses of test material and have at least one post treatment efficacy measurement will be included in the analysis. Comparisons between treatments will be assessed using an *** with factors of ***, ***and *** and with ***. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant at ***, then the test will proceed to the *** also at the ***.”
To: “All subjects who are randomized, take one or more doses of test material and have at least one post treatment efficacy measurement will be included in the analysis. Comparisons between treatments will be assessed using an *** with factors of ***, *** and *** and with *** for percent weight loss, and *** for percentage of subjects with at least 5% weight loss. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant at *** for both co-primary endpoints, then the test will proceed to the *** also at the ***.” |
|
|
|
|
|
|
|
Background |
|
*** |
|
Change: “Obesity is associated with numerous co-morbidities including dyslipidemia, coronary artery disease (CAD), hypertension, stroke and type 2 diabetes.(7), (9) Epidemiological data indicate that obesity is associated with |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
increased mortality,(10) and a recent study of over 500,000 individuals concluded that excess body weight during midlife, including overweight, was associated with an increased risk of death.(11)”
To: “Obesity is associated with numerous co-morbidities including dyslipidemia, coronary artery disease (CAD), hypertension, stroke and type 2 diabetes.(7), (9) Epidemiological data indicate that obesity is associated with increased mortality,(10) and a recent study of over 500,000 individuals concluded that excess body weight during midlife, was associated with an increased risk of death.(11)” |
|
|
|
|
|
|
|
Background |
|
*** |
|
Change: “Topiramate, a sulfamate-substituted monosaccharide, is an anticonvulsant agent indicated as adjunctive therapy for partial onset seizures, primary generalized tonic-clonic seizures, seizures associated with Lennox-Gastaut syndrome and for migraine headache prophylaxis.(5) The recommended total daily dose of topiramate for treatment of seizures in adults is *** administered in two divided doses; ***. Topiramate is known to ***.(5) Recent clinical studies have shown that topiramate can promote weight loss in ***.(16),(17),(18) However, the exact mechanism by which topiramate exerts its anorectic effect is unknown although it is postulated to ***.”
To: “Topiramate, a sulfamate-substituted monosaccharide, is an anticonvulsant agent indicated as adjunctive therapy for partial onset seizures, primary generalized tonic-clonic seizures, seizures associated with Lennox-Gastaut syndrome and for migraine headache prophylaxis.(5) The recommended total daily dose of topiramate for treatment of seizures in adults is ***administered in two divided doses; ***. Topiramate is known to ***.(5) Recent clinical studies have shown that topiramate can promote weight loss in ***.(16),(17),(18) However, the exact mechanism by which topiramate exerts its anorectic effect is unknown although it is postulated to ***.” |
|
|
|
|
|
|
|
Exclusion Criteria #13 |
|
*** |
|
Change: “Diagnosis of type 1 diabetes or history of use of any antidiabetic medication other than metformin;”
To: “Diagnosis of type 1 diabetes or use of any antidiabetic medication other than metformin within the past month;” |
|
|
|
|
|
|
|
Titration (Visit 3) |
|
*** |
|
Change: “Review the study medication used since Visit 2; assess treatment compliance and perform drug accountability. Re-dispense the card to the subject for use during weeks 3 and 4;”
To: “Review the study medication used since Visit 2; assess treatment compliance and perform drug accountability. Re-dispense the titration card to the subject for use during weeks 3 and 4;” |
|
|
|
|
|
|
|
Oral Glucose Tolerance Test |
|
*** |
|
Change: “An oral glucose tolerance test (OGTT) will be obtained at Visit 1a (after results from Visit 1 indicate the subject may be eligible for participation), at Visit 4 (week 4) and at end of treatment (Visit 17 or study withdrawal).”
To: “An oral glucose tolerance test (OGTT) will be obtained at Visit 1a (after results from Visit 1 indicate the subject may be eligible for participation), at Visit 4 (week 4) and at end of treatment (Visit 17 or study withdrawal) for all subjects.” |
|
|
|
|
|
|
|
Analysis of Primary Endpoints |
|
*** |
|
Change: The primary calculated endpoints for the trial are based on the percent weight loss at week 56 calculated as *** and the percentage of subjects achieving at least 5% weight loss at week 56.
The intent to treat (ITT) population (Section 9.2.1.3) is the primary subject population. For subjects who discontinue treatment prior to trial completion, every attempt will be made to have them return to the clinic at week 56 for a final weight assessment, regardless of when they discontinued treatment. For subjects |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
who ***.
Comparisons between treatments will be assessed using an *** with factors of ***, *** and ***, and with ***. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant for the primary end point at ***, then the test will proceed to the *** also at ***.”
To: “The primary calculated endpoints for the trial are the percent weight loss at week 56 calculated as *** and the percentage of subjects achieving at least 5% weight loss at week 56.
The intent to treat (ITT) population (Section 9.2.1.3) is the primary analysis population. For subjects who discontinue treatment prior to trial completion, every attempt will be made to have them return to the clinic at week 56 for a final weight assessment, regardless of when they discontinued treatment. For subjects who ***.
Comparisons between treatments for percent weight loss will be assessed using an *** with factors of ***. Comparisons between treatments for the percentage of subjects achieving at least 5% weight loss will be assessed by *** with ***, ***, and *** and ***. A step-down multiple comparison procedure will be used to compare each dose group with ***. That is, comparison with *** will start at the ***. If the statistical test is significant for both co-primary end points at ***, then the test will proceed to the *** also at ***.” |
|
|
|
|
|
|
|
Analysis of Secondary Endpoints |
|
*** |
|
Change: “The difference in absolute weight reduction and the difference in waist reduction at week 56 from baseline will also be compared using the same *** as the primary end point. *** with ***, *** and *** and ***, will be used to compare the probability to reach 5% and 10% body weight reduction between baseline and at week 56 between treatment groups.”
To: “The difference in absolute weight reduction and the difference in waist reduction at week 56 from baseline will also be compared using the same *** as the primary end point. *** with ***, *** and *** and ***, will be used to compare the probability of reaching 5% and 10% body weight reduction from baseline to week 56 between treatment groups.” |
|
|
|
|
|
|
|
Adverse Events |
|
*** |
|
Change: “Adverse events will be coded using a MedDRA coding dictionary. The number and percentage of subjects who reported at least one adverse event in each organ class and preferred term category, and the total number and percentage of subjects with any AE over all organ system will be summarized by treatment group.
Subsets of AEs that are considered serious or required discontinuation of the study medication will be listed by subject and present separately.”
To: “Adverse events will be coded using a MedDRA coding dictionary. The number and percentage of subjects who reported at least one adverse event in each system organ class and preferred term category, and the total number and percentage of subjects with any AE overall system organ classes will be summarized by treatment group.
Subsets of AEs that are considered serious or required discontinuation of the study medication will be listed by subject and presented separately.” |
|
|
|
|
|
|
|
Interim Analysis |
|
*** |
|
Change: “An interim analysis may be conducted once approximately half of the study subjects have completed the intended course of treatment to provide |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
information for the planning of future clinical trials that may be designed or commenced prior to the completion of this trial. No changes will be made to the conduct of the present study, including premature termination or increased sample size, based on the results of this interim analysis and no statistical adjustments for this interim analysis, if conducted.”
To: “An interim analysis may be conducted once approximately half of the study subjects have completed the intended course of treatment to provide information for the planning of future clinical trials that may be designed or commenced prior to the completion of this trial. No changes will be made to the conduct of the present study, including premature termination or increased sample size, based on the results of this interim analysis and no adjustments to the significance level for the primary hypothesis will be made if this interim analysis is conducted.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “No *** will be employed for this study.”
To: “***” |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Protocol Title: A Phase III Randomized, Double-Blind, Placebo controlled Multicenter Study to Determine the Safety and Efficacy of VI-0521 in the Treatment of Obesity in Adults with Obesity-Related Co-Morbid Conditions
Protocol Number: OB-303
Indicate any amendments to this protocol by writing the Amendment Number and the Date of the Amendment in the space below. This page will be used to track the original protocol and its amendments. This is the original protocol if no amendment number is listed below.
Amendment Number 1
Date of Amendment: ***
(22 items)
This protocol amendment is being implemented to add a co-primary endpoint to the study analysis section, to add specific guidance for the implementation or adjustment of concomitant antidiabetic therapies based on blood glucose values observed during the course of the study, and to add additional laboratory tests to the schedule of evaluations. The definition of child-bearing potential has also been clarified, and miscellaneous typographical errors have been corrected. These changes are being made at the request of the FDA, and are not anticipated to significantly affect the risk to study subjects.
|
Section and/or Item |
|
Protocol |
|
Amendment 1, *** |
|
Protocol Synopsis: Study Subjects |
|
*** |
|
Change: “The study population will consist of *** (BMI > ***and < ***) adults < 70 years of age with *** and at least *** of the following obesity-related comorbid conditions:”
To: “The study population will consist of *** (BMI > *** and < ***) adults < 70 years of age with at least *** of the following obesity-related comorbid conditions:” |
|
|
|
|
|
|
|
Protocol Synopsis: Efficacy Endpoints |
|
*** |
|
Change: “The primary endpoint is the difference between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56). Percent weight loss will be calculated as ***.”
To: “The primary endpoints are the differences between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), and percent of subjects with weight loss of 5% or more at end of treatment (week 56). Percent weight loss will be calculated as ***. |
|
|
|
|
|
|
|
Protocol Synopsis: Efficacy Endpoints |
|
*** |
|
Change: “The difference in percent of subjects who achieve a reduction in total body weight of at least 5% and at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups; and
To: “The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups; and |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
List of Abbreviations |
|
*** |
|
Added: “I-CAM: Intercellular Cell Adhesion Molecule”, “PAI-1: Platelet Activator Inhibitor 1”, “RBP-4: Retinol Binding Protein 4” and “V-CAM: Vascular Cell Adhesion Molecule” |
|
|
|
|
|
|
|
Introduction |
|
*** |
|
Change: “The present study is being conducted to evaluate the combined use of phentermine and topiramate at doses of 7.5 mg phentermine/46 mg topiramate and 15 mg phentermine/92 mg topiramate in the treatment of obesity in adult subjects with body mass index (BMI) greater than or equal to ***.”
To: “The present study is being conducted to evaluate the combined use of phentermine and topiramate at doses of 7.5 mg phentermine/46 mg topiramate and 15 mg phentermine/92 mg topiramate in the treatment of obesity in adult subjects.” |
|
|
|
|
|
|
|
Trial Design |
|
*** |
|
Change: “The primary endpoint is the difference between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56). Percent weight loss will be calculated as ***.
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) for *** and *** groups.
· The difference in percent of subjects who achieve a reduction in total body weight of at least 5% and at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups.”
To: “The primary endpoints are differences between *** and *** groups in mean percent loss of baseline body weight, determined by weight at randomization (baseline) and weight at end of treatment (week 56), and percent of subjects with weight loss of 5% or more at end of treatment (week 56). Percent weight loss will be calculated as ***.
Secondary efficacy endpoints are:
· The difference in absolute weight loss between randomization (baseline) and end of treatment (week 56) for active and placebo groups.
· The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups.” |
|
|
|
|
|
|
|
Inclusion Criteria |
|
*** |
|
Change: “Have a BMI >*** and <***;”
To: “Have a BMI >*** and <*** (for diabetic subjects, there is no lower limit on BMI for study inclusion);” |
|
|
|
|
|
|
|
Inclusion Criteria |
|
*** |
|
Change: “If females of child-bearing potential, be using adequate contraception, defined as double barrier methods, stable hormonal contraception plus single barrier method or tubal ligation. Females are considered to be of child-bearing potential unless they have undergone a hysterectomy or bilateral oophorectomy, have a documented follicle stimulating hormone level >40 IU/L or are 55 years of age or greater and have experienced cessation of menses for at least 1 year;
To: “If females of child-bearing potential, be using adequate contraception, defined as double barrier methods, stable hormonal contraception plus single barrier method or tubal ligation. Females are considered to be of child-bearing potential unless they have undergone a hysterectomy or bilateral oophorectomy, are 55 years of age or greater and have experienced spontaneous cessation of |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
menses for at least 1 year, or have a documented follicle stimulating hormone level >40 IU/L;” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “When *** is not appropriate or when *** may be required due to events unrelated to subject treatment, subjects may ***. If dosing has been ***, a new titration kit should be ordered through *** to ***.”
To: “When *** is not appropriate or when *** may be required due to events unrelated to subject treatment, subjects may ***. *** are possible with agreement from the medical monitor. All subjects undergoing *** for *** may be *** based on discretion of the PI. If dosing has been ***, a new titration kit should be ordered through *** to ***.” |
|
|
|
|
|
|
|
Treatment of Diabetes |
|
*** |
|
Change: “Subjects with consistently elevated fasting blood glucose values should ***. Subjects whose fasting blood glucose ***, or whose blood glucose cannot be adequately controlled with the concomitant treatments allowed in this trial should be discontinued from study treatment and referred back to their primary healthcare provider for additional glycemic management. Subjects may continue attending study visits ***.”
To: “Subjects with consistently elevated fasting blood glucose values should ***. For this study, actionable levels for glucose values are ***. Subjects who note *** fasting glucose values that exceed these thresholds in daily glucose monitoring logs during the week prior to a study visit would be considered appropriate for concomitant medication adjustment. Subjects whose fasting blood glucose ***, or whose blood glucose cannot be adequately controlled with the concomitant treatments allowed in this trial should be discontinued from study treatment and referred back to their primary healthcare provider for additional glycemic management.” |
|
|
|
|
|
|
|
Randomization (Visit 2) |
|
*** |
|
Change: “Assess adverse events (including eye symptoms), if any;”
To: “Assess adverse events (including eye symptoms and ***), if any;” |
|
|
|
|
|
|
|
Titration (Visit 3) |
|
*** |
|
Change: “Assess adverse events (including eye symptoms), if any;”
To: “Assess adverse events (including eye symptoms and ***), if any;” |
|
|
|
|
|
|
|
Treatment (Visits 4 through 16) |
|
*** |
|
Change: “Assess adverse events (including eye symptoms), if any;”
To: “Assess adverse events (including eye symptoms and ***), if any;” |
|
|
|
|
|
|
|
End of Treatment (Visit 17 or Withdrawal from Study) |
|
*** |
|
Change: “Assess adverse events (including eye symptoms), if any;”
To: “Assess adverse events (including eye symptoms and ***), if any;” |
|
|
|
|
|
|
|
Biomarkers |
|
*** |
|
Change: “Appropriate blood and urine biomarkers [C-reactive protein, microalbumin (urine) and creatinine (urine)] will be evaluated at screening (visit 1a), visit 10 (week 28) and end of treatment (visit 17 or study withdrawal).”
To: “Appropriate blood and urine biomarkers [C-reactive protein, microalbumin (urine) and creatinine (urine)] will be evaluated at screening (visit 1a), visit 10 (week 28) and end of treatment (visit 17 or study withdrawal). Additionally, adiponectin, intercellular cell adhesion molecule (I-CAM), vascular cell adhesion molecule (V-CAM), retinal binding protein 4 (RBP-4), platelet activator inhibitor 1 (PAI-1) and fibrinogen will be evaluated at screening (visit 1a) and end of treatment (visit 17 or study withdrawal). With the exception of urine microalbumin and creatinine, all post-screening biomarker results will be blinded to investigators and sponsor.” |
|
|
|
|
|
|
|
Eliciting Adverse Event Information |
|
*** |
|
Change: “The following sections describe additional requirements to ensure ***.” |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
|
|
|
|
To: “The following sections describe additional requirements to ensure ***.” |
|
|
|
|
|
|
|
*** |
|
*** |
|
Change: “Should this additional assessment indicate the presence ***”
To: “Should this additional assessment indicate the presence of ***” |
|
|
|
|
|
|
|
Hypoglycemia |
|
*** |
|
New Section added and new Table:
“Guidelines for the reporting of adverse events based on results obtained in *** maintained by *** are summarized in Table 2.
Table 3. Guidelines for adverse event reports based on ***” |
|
|
|
|
|
|
|
Analysis of Primary Endpoints |
|
*** |
|
Change: “***
The primary calculated endpoint for the trial is based on the percent weight loss at week 56 calculated as ***.”
To: “***
The primary calculated endpoints for the trial are based on the percent weight loss at week 56 calculated as *** and the percentage of subjects achieving at least 5% weight loss at week 56.” |
|
|
|
|
|
|
|
Analysis of Secondary Endpoint |
|
*** |
|
Change: “The difference in percent of subjects who achieve a reduction in total body weight of at least 5% and at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups.”
To: “The difference in percent of subjects who achieve a reduction in total body weight of at least 10% between randomization (baseline) and end of treatment (week 56) for *** and *** groups.” |
|
|
|
|
|
|
|
Interim Analysis |
|
*** |
|
Change: “No changes will be made to the conduct of the present study, including premature termination or”
To: “No changes will be made to the conduct of the present study, including premature termination or increased sample size, based on the results of this interim analysis and no statistical adjustments for this interim analysis, if conducted.” |
|
|
|
|
|
|
|
Appendix 1 |
|
*** |
|
Change: “***”
To: “***” |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #03
Appendix 2 — Scope of Work
VIVUS, Inc.
Qnexa OB-303
PROJECT OVERVIEW
▇▇-▇▇▇
▇▇▇ ▇▇-▇▇▇ trial is a phase III randomized, double blind, placebo controlled multicenter study to determine the safety and efficacy of VI-0521 in the treatment of obesity in adults with obesity-related comorbid conditions.
PROJECT TEAM
Study Management
The overall management of the study will be the responsibility of the Senior Clinical Trial Manager (CTM). The Senior CTM will oversee and coordinate the management of the study as well as oversee the study specific CTM. This oversight will ensure consistency and allow VIVUS Study Management to have one primary contact for the Qnexa program. The Medpace CTM assigned to OB-303 will work closely with the VIVUS Clinical Leader and Medpace Medical Expert to address protocol questions and interpretations while maintaining close oversight of study-related processes and documents. The OB-303 CTM will supervise all Clinical Research Associates (CRAs) and Project Coordinators assigned to the project.
The Project Coordinators will be responsible for day-to-day study management functions, including the generation of status reports, organization of supplies, generation and compilation of newsletters, and input of all study information into the ClinTrak® Study Management System, a web-based, proprietary research management system designed by Medpace. The Project Coordinators will organize teleconferences and team meetings, including the compilation of agendas and meeting minutes.
The Study Start-Up Manager and Study Start-Up Coordinators will work closely with the CTM and Project Coordinators to ensure sites become active in the most time effective manner.
The Medpace Contracts Attorney will be responsible for the execution of Investigator contracts (upon VIVUS defined process). The Contracts Attorney will work closely with the Start-Up Manager and Medpace CTM to ensure contracts are executed in a timely manner.
The Medpace Medical Expert assigned to this project will work closely with the VIVUS Clinical Leader . The Medpace Medical Expert will assist with protocol design and medical interpretation of entry criteria and adverse events (AEs). The Medical Expert will also be involved in the training of CRAs and other staff members participating in the
|
CONFIDENTIAL |
|
September 7, 2007 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
project. The Medical Expert will review and approve the coding of concomitant medications, medical histories, AEs, and will provide the medical context for the statistical analysis and medical writing.
The Medical Expert will assist in the review of the protocol, train Medpace personnel internally as to the background of the study compound and design of the study, participate in the project teleconferences and meetings, work hand-in-hand with the OB-303 CTM, and have heavy involvement in the clinical study report. The Medical Expert’s role and decision making rights are dictated by VIVUS (e.g. inclusion/exclusion of patients, discussions with Investigators about withdrawing a patient, etc.). This decision making power often times reduces the oversight needed by the sponsor. For questions the OB-303 CTM is not comfortable answering, she will contact the Medical Expert for guidance. Obviously, VIVUS will be involved in study oversight based on pre-defined terms with the VIVUS Clinical Development Team. The Medpace Medical Expert is available 24 hours a day, 7 days a week via the Medpace Project Helpline.
Clinical Monitoring
Medpace operates in North America with a primarily centralized monitoring team of over 140 CRAs to promote greater standardization, cohesiveness, support, and stability. Each of the Medpace CRAs assigned to this project have monitoring experience and strong clinical backgrounds.
Clinical Safety
The Clinical Safety will be managed by VIVUS or its designee. VIVUS Clinical Leader to be involved with casualty assignment for all Serious Adverse Events (SAEs).
Data Management
A Data Manager will serve as the primary contact for the Data Management team. Data Coordinators will be involved in the day-to-day operations and report issues to the Data Manager. Data Entry Specialists and Database Programmers will also be utilized.
Biometrics
Key members of our Biometrics team include Biostatisticians and Statistical Analysts. The Biostatistician assigned to this project will develop the analysis plan and coordinate biometrics activities. The Lead Statistical Analyst will work closely with the Biostatistician to ensure a clear understanding of the analysis plan and communicate any programming issues that may arise.
Medical Writing
The Medical Writing team works in collaboration with the Medical Experts to prepare research reports meeting International Conference on Harmonisation (ICH) and Sponsor guidelines. All Medpace Medical Writers have extensive experience in regulatory
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
submission preparation. The Medical Writing team is actively involved throughout the conduct of the trial.
Team Organization Chart
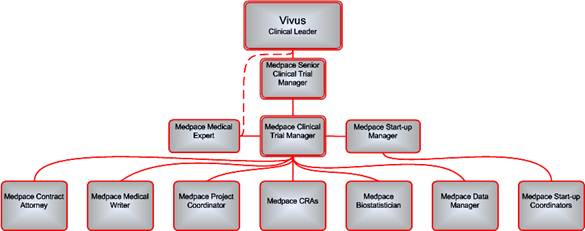
PROJECT START-UP
Protocol
VIVUS will prepare the protocol. Medpace will review the protocol and provide comments before it is finalized.
Case Report Forms
Medpace will design the electronic case report forms (eCRFs) for the trial, including completion instructions, according to the final protocols and the Medpace template. VIVUS must review and approve the eCRFs before they are finalized.
Project Initiation
Prior to the study site initiation visits, a project kickoff meeting will be held at Medpace involving Medpace and VIVUS personnel to review the study protocol, eCRFs, and overall project coordination. Medpace project team members and VIVUS personnel will participate in this meeting.
Interactive Voice Response System
Medpace will provide a customized (study-specific) interactive voice response system (IVRS) to provide patient randomization, and drug management. The Medpace IVRS is a proprietary in-house developed system. The system provides both voice and web access and has been developed in conjunction with our web based Clintrak® system providing
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
seamless functionality throughout the conduct of the study. The VIVUS Team (no limit applied to number of team members) will have access to review reports within the IVRS. Medpace will perform the User Acceptance Testing (UAT) for each site.
The IVRS will include:
· Subject Screens/Screen Failures;
· Subject randomization;
· Patient visit tracking;
· Inventory management (site supply set-up, initial bulk supply, and resupply of one additional shipment per patient);
· Notifications of site shipments;
· Confirmation of receipt of shipments; and
· Customized reports.
VIVUS will review IVRS and approve system prior to finalization.
Study Medication Supply and Storage
VIVUS will be responsible for the supply, packaging, labeling, storage, and destruction of study medication. Distribution of the study medication will be tracked and initiated via the Medpace IVRS. Study medication accountability procedures will follow Medpace standard operating procedures (SOPs) and utilize a study medication accountability log that has been approved by VIVUS.
Recruitment Oversight Plan
The Medpace CTM, in collaboration with VIVUS, will develop a recruitment oversight plan. Medpace understands the importance of rapid recruitment and the necessity to keep patients in the trial until completion. Medpace will develop processes prior to study initiation to ensure recruitment is efficient and retention of patients in the study is maximized. The plan will include details on:
· Initial collection of essential documents;
· Patient recruitment (including tools, site-specific plans, contingencies, etc.); and
· Patient recruitment tracking reports.
The tools noted above include:
· Inclusion/exclusion cards;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Patient emergency cards;
· Enrollment tracking forms;
· Laminated patient visit schedule;
· Pocket protocol; and
· Advertising tools (e.g., posters, table tents, language for newspaper/radio advertisements, etc.).
Medpace understands each site has a different experience and approach to patient recruitment. The Medpace CTM and Medpace CRAs will work with personnel from each site to help maximize their efforts. The project team maintains frequent contact during the active recruitment phase to assess each site’s activity and offer assistance when needed. Medpace acknowledges the importance of site recognition and offers various incentives throughout the recruitment period. Examples of site incentives are “Site of the Month” awards and weekly faxes displaying each site’s activity (often included in the newsletter as well). The time associated with these types of incentives are inclusive of the budget. However, often times, monetary incentives are built into the Investigator’s grant to encourage rapid essential document collection and/or timely recruitment.
Patient retention is vital to the success of a trial. The Medpace CTM and the Medpace CRAs will work with the sites to understand the needs and motivations for patients to remain in the study. They will help educate the sites on the importance of “customer service” and “patient satisfaction” as elements that ensure continued patient participation. Examples of patient retention methods include quarterly patient newsletters, ideas for site customer service, and tokens of appreciation for patients.
Site Selection and Pre-study Visits
VIVUS, in conjunction with Medpace, will identify qualified Investigators. VIVUS and Medpace will also work together to provide and negotiate Confidentiality Disclosure Agreements (CDAs) as well as create and evaluate site questionnaires. In not knowing the number of pre-study visits Medpace will be required to conduct, a unit price for a pre-study visit has been provided in the budget.
Pre-study visits will be conducted consistent with Medpace SOPs. Medpace will provide a pre-study visit report to VIVUS within 10 business days of each visit.
These visits will include, but are not limited to:
· Determining whether or not the site has clinical staff of appropriate education, training, and experience to manage the study and have sufficient capacity to perform the required tasks;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Determining whether or not the site has appropriate facilities to conduct the study;
· Determining there are no competing studies that will conflict with patient enrollment and that the site has sufficient patients and processes to enroll patients in the time identified for patient recruitment; and
· Determining whether or not the site has appropriate resources and procedures to maintain appropriate records according to FDA requirements.
Study Start-up Team
The efficient start-up of the *** study sites for OB-303 will have a significant effect on patient recruitment time. Medpace will utilize its Study Start-up team for additional support during the initial phase of the trials to expedite the overall study start-up for each site.
The Study Start-up team works directly with the CTM and the Project Coordinators. The team is comprised of a Study Start-up Manager and several experienced Study Start-up Coordinators. The team is responsible for many of the key start-up activities, including:
· Submission to the central IRB;
· Coordination and tracking of essential documents packages for each site;
· Investigator meeting presentations and binders; and
· Site tools.
Central Laboratory Selection
Medpace Reference Laboratories (MRL) will be utilized for processing the clinical laboratory samples. MRL is committed to providing comprehensive laboratory services of the highest quality to the pharmaceutical and biotechnology industries.
Investigators’ Meeting
An Investigators’ Meeting will be held for the OB-303 study. VIVUS will arrange the meeting (including contracting with a third-party vendor) and Medpace will prepare the meeting materials, including preparation and distribution of binders. The Medpace OB-303 Team will attend the meeting. VIVUS will open the meeting and Medpace will present on the topics delegated by VIVUS. The meeting minutes will be prepared by Medpace, reviewed and approved by VIVUS, and distributed to the study sites by Medpace. The preparation of the meeting minutes is optional; however, is included in the budget. Medpace assumes the Investigators’ Meeting will serve as the initiation visit for the Investigators. Therefore, the budget reflects 20% of the sites will require an initiation visit (for those unable to attend the Investigators’ Meeting).
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Clinical Trial Agreements for Sites, Central Laboratory, and EDC Vendor
Medpace will prepare and provide sample clinical trial agreements (including budgets) for the study sites. VIVUS will review and approve the final draft versions of the clinical trial agreements. The agreements will be distributed and negotiated with each site by Medpace (with final approval by VIVUS). Medpace will make payments to the clinical sites according to the VIVUS-approved schedule. All payments for sites, Medpace Reference Laboratories, and Phase Forward will be made electronically to Medpace within seven days of invoice receipt. If electronic payment exceeds or falls below actual costs, VIVUS will adjust payment based on the prior month’s payment reconciliation. Investigator payment invoices will include the following detail:
· Clinical study number;
· PI or Site #;
· Patient ID;
· Amounts paid per visit;
· Total amount earned to date;
· Prior payments; and
· Current payment amount.
Institutional Review Board and Initial Essential Documents Packages
Medpace will select a central Institutional Review Board (IRB) and coordinate the initial submissions to the IRB. VIVUS must approve the central IRB selected. Medpace will be responsible for payments to the central IRB utilizing funds provided in the same manner as described above.
A study-specific, prototype informed consent form (ICF) will be designed by Medpace. The ICFs will be reviewed and approved by VIVUS. Medpace will distribute the ICFs to the Central IRB. Medpace will be responsible for negotiating changes to the informed consents with the central IRB.
Deviations from the VIVUS template must be brought to the attention of the VIVUS Clinical Leader who will facilitate VIVUS legal review and approval, if required.
All components of the Initial Essential Documents Package will be collected, tracked, and maintained by Medpace according to Medpace SOPs. The Medpace Study Start-up team will review all documents, negotiate any changes with study site personnel, and correct any errors. The Initial Essential Document Package includes the following:
· Signed protocol signature pages;
· Financial disclosure questionnaires (FDQs) (template to be provided by VIVUS, Medpace to collect the forms);
· Clinical study agreement (includes study budget);
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· FDA Form 1572;
· Laboratory certifications and reference values;
· Curricula vitae for all Investigators;
· IRB approval of the protocol (and any amendments), the informed consent and sponsor approved advertisements; and
· Qualification of IRB members.
The FDQs shall apply throughout the entire term of the study and for one year following last patient last visit (LPLV). If there is any change in the accuracy of a particular site’s FDQ during that time period, that site will be responsible for notifying Medpace of the change. Medpace will send a fax to all sites once the study has ended reminding them of their responsibilities (one of which includes notifying Medpace of any FDQ changes). If a site notifies Medpace of a change in staff from LPLV to one year after LPLV, Medpace will collect an updated FDQ and forward on to VIVUS. Costs associated with this task are included in the budget.
Site Initiation Visits
Site initiation visits will be conducted by the Medpace CRAs consistent with Medpace SOPs. For purposes of this proposal, it is assumed that the Investigators’ Meeting will serve as the initiation visit and only 20% of the sites for each study will require separate initiation visits. Typically, if the Investigator Meeting is considered the initiation visit, the CRA will contact the site via phone to review study procedures and the CRA’s first routine monitoring visit will occur shortly (can be defined as 1 or 2 weeks) after a patient is screened for the trial. During this visit, the CRA will review bullets 2-5 below. A site initiation visit report will be completed and forwarded to VIVUS within 10 business days of the visit. These visits will include, but are not limited to, the following tasks:
· Train site and applicable study personnel on the protocol and study procedures;
· Ensure the site has received all study supplies required for the conduct of the study, including study medication and access to eCRFs;
· Provide and review the Trial Master File binder. Medpace CRAs will provide instruction to the site personnel on the organization and maintenance of the documents in the binder;
· Review study medication accountability procedures;
· Provide eCRF completion instructions; and
· Explain the serious adverse event (SAE) reporting procedures.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
CLINICAL OPERATIONS
Monitoring Data Review Guidelines
The Medpace Lead CRA in collaboration with the project team members will develop a project-specific Monitoring Plan for the study. This plan will include detailed interpretations of study expectations for the CRAs assigned to the study. Issues are discussed and updated on an ongoing basis throughout the project. Medpace will request that VIVUS approve the initial document and then re-approve the document on a quarterly basis.
Routine Clinical Monitoring Visits
Medpace will conduct routine monitoring visits at each site consistent with Medpace SOPs. The frequency of the visits will be determined by the site’s activity, but will be conducted on average every four to six weeks. Visits at the beginning and end of the study may be more frequent based on the needs of the study, including, but not limited to, recruitment, quality data and study close-out activities. Based on recruitment being very rapid, the first routine visit will be performed within 2 weeks after the first two patients are screened at the site. The CRA will perform 100% source documentation. In addition, data queries will be resolved during the visits, eCRF changes will be verified, and supporting documentation for SAEs will be obtained. The Medpace CRA will verify all laboratory samples have been obtained according to guidelines and the results are available in the patient’s source documents. A monitoring visit report will be forwarded to VIVUS within 10 business days of the visit. VIVUS will be notified of any significant issues by phone within one business day.
The following tasks will also be performed:
· Train any new site personnel and review study issues with applicable site personnel;
· Ensure the site has sufficient study supplies (including study medication);
· Ensure the site is entering eligible patients into the study in a timely manner, and notify the Medpace Study Manager immediately of any problems;
· Detect any significant compliance or other issues and notify the Medpace Study Manager by phone, within one business day of the monitoring visit;
· Confirm the Trial Master File is complete and current, and the site is complying with applicable regulations and the protocol. VIVUS will be notified immediately of any significant deviations;
· Ensure the site is completing eCRFs in a timely manner;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Ensure all completed eCRFs are reviewed, verified, corrected, and transmitted to Medpace;
· Review eCRFs for accuracy and protocol adherence;
· Verify study medication dispensing, compliance, and accountability for each patient; and
· Ensure the Investigator reported all SAEs to Medpace and the applicable IRB.
Medpace will provide a follow-up letter to the study site after each visit. The letter will include, but will not be limited to, the following:
· Important findings during the visit;
· Recommendations of corrective actions to be taken by the site; and
· Follow-up information regarding questions asked during the visit.
The Monitoring Visit Reports with all attachments including follow-up letters, will be available for view through the Medpace web based Clintrak® Study Management system within 10 days of the monitoring visit.
In-house Clinical Monitoring Activities
Investigators will be contacted on a regular basis (every week during the active recruitment period and between monitoring visits) to ensure progress at the study site. The CRA will take the opportunity to review enrollment, answer protocol-related questions, discuss eCRF completion issues, obtain information regarding AEs, and ensure the site continues to be committed to the completion of the study in a timely manner and according to the protocol.
Telephone contacts will be entered in the Medpace ClinTrak Study Management system. Contacts requiring urgent attention will be relayed to VIVUS immediately and will be resolved in collaboration with the Medpace CTM and the VIVUS Clinical Leader.
Withdrawals Due to Adverse Events
Withdrawals due to AEs will be tracked and reconciled with the eCRF database on an ongoing basis by Medpace. The CRA will be responsible for reporting withdrawals due to AEs to VIVUS using the monitoring visit report.
Medpace will write narratives for all withdrawals due to AEs, for use in the clinical trial study report.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Status Reporting
The Medpace Senior CTM will serve as the central channel for communication between Medpace and VIVUS. The OB-303 CTM will work in conjunction with the clinical monitoring group to track study progress and report to VIVUS on a weekly basis. In addition, the OB-303 CTM will be responsible for overall management of site information, overseeing the status of Investigator contracts, direct supervision of CRAs, tracking of enrollment information, and distribution of study supplies. The Medpace OB-303 CTM will be the primary contact for the sites to address protocol interpretations and inclusion/exclusion criteria. All protocol-related issues will be recorded in an ongoing document to ensure consistency. The CTM is available 24 hours a day, 7 days a week via the Medpace Project Helpline.
Medpace will develop a communication plan at the start-up of the study. The Senior CTM will work with the VIVUS project team to define details regarding study communication, including status reporting and team conference calls. Medpace typically delivers status reports on a predetermined day (and time) of the week, which is established around the weekly team calls so that the information can be discussed. Utilization of IVRS will also allow the project team to review patient status on a real time basis.
The Medpace CTM will collaborate with the Medpace Medical Expert and the VIVUS Clinical Leader to address any questions that may arise. The ClinTrak Study Management databases will serve as the primary source of project status information and will allow the Medpace CTM to report on any aspect of the study. The databases are updated on a real-time basis, providing accurate and up-to-date information.
Elements of ClinTrak include:
· Phone contacts;
· Monitoring visit reports;
· Patient status including details on withdrawals during the treatment phase;
· Study supplies; and
· Protocol deviations.
Medpace will provide weekly status reports via a secure project website, to include the following status by site:
· Number of patients screened;
· Number of screen failures;
· Number of patients randomized;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Number of patients dropped with drop rates; and
· Number of patients completed.
In addition, monthly reports will be provided, to include the following:
· Monitoring visits scheduled; and
· Monitoring visits completed.
Data Management status reports will be provided monthly via a secure website. These reports will include the following:
· Cumulative and interval eCRF status by site (including number of eCRFs transmitted and cleaned);
· Cumulative and interval patient status by site (including number of patients ongoing, completed, and early terminations) based on eCRF data in-house; and
· Cumulative and interval query status by site (including number of queries issued and days outstanding).
Project Website
Medpace will develop a secure OB-303 website that will be available to all project team members and site personnel. The website will include information and tools relevant to the study, such as status reports, meeting agendas and minutes, newsletters, monitor visit status, and the project timeline. Access is controlled by the type of user. Access to the tabs (sections) on the websites are controlled by the user type so that sites can have access to the section specifically designed for site access. Medpace can set up an automatic notification process of updates to the user email accounts. Clinical sites will have access only to parts of the website that pertain to their function. They will have the ability to receive study information and download study-related forms.
Team Meetings
VIVUS and Medpace
Medpace assumes that one face-to-face meeting, other than the Investigators’ meeting and kickoff meeting will take place for the OB-303 study at VIVUS.
Weekly teleconferences will be held during the study. The teleconferences will be held to discuss study progress and review project documents (as necessary). The Medpace Project Coordinators will be responsible for preparing and distributing agendas and minutes for each meeting/teleconference.
Additional meetings/teleconferences will be scheduled throughout the project, as needed.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Internal Meetings
Medpace will develop a project-specific internal project development/training program for all project team members. Included in this program will be the following:
· Protocol/eCRF review meeting;
· Medical in-services;
· Periodic Monitoring Plan meetings; and
· Periodic project meetings.
Newsletter
Medpace will prepare a 2-4 page full color site monthly newsletter for OB-303 as an additional avenue of communication and training for all site personnel. VIVUS will provide input and approval of the newsletter prior to distribution. Medpace will be responsible for printing and distributing two copies of each newsletter to each site.
Closeout Visits
Medpace will conduct closeout visits at each site consistent with Medpace SOPs after all patients have completed or discontinued from the study at the respective site. This visit may be performed as part of a final routine site monitoring visit. Site closeout visit reports will be forwarded to VIVUS within 10 business days of the visit. The following tasks will be performed:
· Resolve outstanding data queries;
· Ensure all study medication supplies are accounted for and that medication records and unused supplies are returned to VIVUS;
· Ensure the Investigator’s copies of data and source documents are properly stored;
· Ensure the Trial Master File is complete, correct, and properly stored;
· Ensure the Investigator is aware of record retention requirements and other obligations, and a final site status report is sent to the IRB and VIVUS; and
· Instruct the site to update the FDQs for one year after the study is completed.
Site Audits
Site audit visits will be conducted, as deemed necessary, by VIVUS.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Medpace will respond to any audit findings and ensure the proper actions are taken to resolve outstanding issues.
REGULATORY AFFAIRS AND SAFETY REPORTING
Serious Adverse Events
All SAEs will be reported immediately, within 24 hours of discovery or notification of the event, by the clinical study site to VIVUS, or its designee, according to SOPs specified by VIVUS.
VIVUS will be responsible for submitting all immediately reportable SAEs (serious, causally related and unexpected) to the Food and Drug Administration (FDA) in accordance with the current regulations.
If a SAE has occurred at a site, the Medpace CRA will always 100% source document verify the event during the monitoring visit to ensure it has been recorded, documented, and reported appropriately and accurately. In addition, data queries will be resolved during the visits, CRF changes will be verified, and supporting documentation for SAEs will be obtained.
Medpace will provide VIVUS listings of non-serious adverse events from all sites to support filing of the Annual Safety Reports. Medpace will reconcile SAE listings with the AE database.
DATA MANAGEMENT
Data Management activities performed by Medpace will include eCRF tracking, preparation of a data management manual, eCRF review, coding of adverse events and concomitant medications, medical histories, data cleaning/editing, querying, query tracking, final database quality review, and delivery of the final SAS® database. The data cleaning process will be performed on an ongoing basis following Medpace Data Management SOPs. Two data transfers (SAS transport files) will be performed: a test transfer prior to FPFV and a final transfer. A unit price for additional data transfers has been provided in the budget.
Database Development and Data Management Manual
Medpace will design and validate the data entry systems prior to entry of data. A Data Management Manual will be prepared for each study using the Medpace template, and will include the following data management documents:
· Database specifications, based on VIVUS specifications;
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Guidelines for the tracking of eCRFs and data queries;
· Data Management Guidelines, which will include guidelines for reviewing the data, and description of the database edit check specifications to be performed for data cleaning; and
· Description of the database quality control (QC) plan.
The manuals will be reviewed and approved by VIVUS.
Data from the Central Laboratory
Medpace will arrange periodic data transfers from MRL. Medpace will track and reconcile discrepancies between the MRL demographic data and the eCRF database, which are generated during the data cleanup process throughout each project.
Data Entry and Data Querying
Medpace assumes no codable forms will be transmitted for screen failure patients. Data is entered by site personnel that have been trained on the eCRF system. Data will be reviewed according to Medpace Data Management Guidelines and edits. A data query will be generated electronically within the eCRF system. The resolutions/corrections are made by site personnel by changing the data. All changes are recorded in an audit trail. All answered queries are verified/closed by Medpace Data Management. All resolutions/corrections will be performed consistent with Medpace SOPs. The Data Coordinators will work directly with the site personnel in resolving queries.
Coding
Medpace will be responsible for coding adverse events, medical histories, and concomitant medications.
· MedDRA will be used to code adverse events and medical histories. Adverse events and medical histories will be coded to the lowest level term, preferred term, and system organ class.
· WHO DRUG will be used to code concomitant medications. Concomitant medications will be coded to the generic name and anatomic therapeutic class 3. It is assumed by Medpace that VIVUS holds a valid agreement with the Uppsala Monitoring Centre (UMC) for the WHO DRUG dictionary.
All coding will be done on a single version of each coding system (versions to be agreed upon by VIVUS). Medpace will provide the coding dictionaries.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Data Flow Chart
STATISTICAL ANALYSIS
Medpace will develop the data analysis plan (DAP) per the VIVUS/Medpace format and template. VIVUS will review and approve the DAP prior to initiation of programming. Included in the DAP is a detailed statistical methodology and programming specification for all statistical analyses, tables/figures/listings (TFLs), and derived datasets.
Medpace will be responsible for programming and generating all TFLs, according to the Medpace standard analysis validation and quality control procedures. All TFLs will be programmed using SAS (Version 8), according to the VIVUS Programming Standards document. Pre-final TFLs will be generated twice on clean data for format review. Final TFLs will be generated on final data for the clinical study report.
MEDICAL WRITING
The Medpace Medical Writing team works in collaboration with the Medpace Medical Expert to provide a clinical study report according to FDA/ICH guidelines. The preparation of the Integrated Clinical/Statistical Study Report involves three stages of development: (1) the Study Report Shell (SRS), (2) the Pre-Final Study Report (PFSR), and (3) the Final Study Report (FSR).
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
The SRS is prepared after sign-off of the Final DAP. The SRS is created using a template and/or style guide provided by VIVUS, or by utilizing the Medpace standard report template, which adheres to the ICH guideline, “Structure and Content of Clinical Study Reports,” and follows the American Medical Association Manual of Style. The SRS incorporates information from the protocol, amendments, and eCRFs into sections of the report, including but not limited to the study design, study population, treatments administered, and the evaluation schedule. The statistical methods and results sections encompass information derived from the Final DAP. The results sections include mock-up in-text tables and text. The SRS undergoes a complete team review, which includes the Medical Monitor, Statistician, CTM, Data Manager, and other team members, if applicable. After the Medical Writer incorporates all team changes into the SRS, the SRS undergoes Medpace’s comprehensive document QC process. Once all changes from the document QC process are implemented into the SRS, the SRS is forwarded to the VIVUS for review.
Preparation of the PFSR occurs after receipt and implementation of VIVUS comments on the SRS, declaration of a clean database, and completion of Pre-Final Analyses. If necessary, a results review meeting is conducted with key members of the Medpace and VIVUS project team as the Medical Writer begins preparing the PFSR. The PFSR is a complete version of the report without the appendices. The PFSR undergoes a complete team review, which includes the Medical Monitor, Statistician, CTM, Data Manager, and other team members, if applicable. After the Medical Writer incorporates all team changes into the PFSR, the PFSR undergoes Medpace’s comprehensive document QC process. Once all changes from the document QC process are implemented into the PFSR, the PFSR is forwarded to VIVUS for review.
The FSR is prepared once VIVUS’ comments on the PFSR are returned and all requested changes are agreed upon (including the acceptance of final text for all complicated or sensitive sections, which may require sending non-QC’d drafts of the report to VIVUS), and the Final Analyses are completed. The FSR is a complete version of the report including paginated appendices and/or supplements. The FSR undergoes a complete internal QC review and is forwarded to VIVUS for sign-off. The signed cover sheet is returned by VIVUS to verify their acceptance of the FSR.
STUDY CLOSEOUT
At the conclusion of each study, once all deliverables have been met, Medpace will return the original study files to VIVUS. These will include:
· General project administration files (this file includes items such as: Outside Vendor Correspondence, Multiple Site Correspondence (e.g. faxes to all sites), Central IRB Correspondence, Project Specific SOPs and Procedures, Monitoring Plan, Trial Master File, Project Timelines, Newsletters, and Meeting Minutes)
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
· Site files (this file includes site items such as: Essential Documents, Budget, Site Correspondence, Monitoring Visit Reports, Drug shipment documents, Study Supply forms, and Protocol Deviations);
· Final statistical tables and listings with results;
· Data management documentation (this file includes items such as: Database Definition Document, Final eCRFs, External Database Import Specs., Data Management Plan, Coding Reports, Analysis Plan, and Randomization Code); and
· Final clinical study report.
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Assumptions
|
OB-303 |
|
Description |
|
Number of Investigators |
|
*** |
|
Number of Screened Patients |
|
*** |
|
Number of Randomized Patients |
|
*** |
|
Number of Randomized Patients/Site |
|
*** |
|
Duration of Enrollment Period |
|
*** |
|
Duration of Treatment Period |
|
*** |
|
Number of Investigators’ Meetings |
|
*** |
|
Number of Kickoff Meetings (at Medpace) |
|
*** |
|
Number of Sponsor Meetings (at VIVUS) |
|
*** |
|
Number of Conference Calls |
|
*** |
|
Frequency of Conference Calls |
|
*** |
|
Number of Clinical Monitors |
|
*** |
|
Number of Pre-study Visits |
|
*** |
|
Number of Initiation Visits |
|
*** |
|
Number of Routine Monitoring Visits |
|
*** |
|
Number of Closeout Visits |
|
*** |
|
Monitoring Frequency |
|
*** |
|
Number of Newsletters per Site (monthly) |
|
*** |
|
Estimated Number of eCRFs per Completed Patient |
|
*** |
|
Estimated Number of Unique eCRFs per Completed Patient |
|
*** |
|
Total Number of eCRFs |
|
*** |
|
Estimated Number of AE Codes Per Patient |
|
*** |
|
Estimated Number of Medical History Codes Per Patient |
|
*** |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
Estimated Number of Concomitant Medications Codes Per Patient |
|
*** |
|
Estimated Number of Queries Per Patient |
|
*** |
|
External Data Sources |
|
*** |
|
Data Transfers from Medpace to VIVUS |
|
*** |
|
Number of Raw Listings |
|
*** |
|
Number of Unique TFs |
|
*** |
|
Number of Version TFs |
|
*** |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #03
Appendix 3 — Project Schedule
VIVUS, Inc.
Qnexa ▇▇-▇▇▇
▇▇▇▇▇▇▇▇▇▇
|
▇▇-▇▇▇ |
|
Date |
|
Medpace Begins Work |
|
*** |
|
Protocol Finalized |
|
*** |
|
First Patient First Visit |
|
*** |
|
Last Patient First Visit |
|
*** |
|
Last Patient Last Visit |
|
*** |
|
Final Database Lock |
|
*** |
|
Final TFLs Available |
|
*** |
|
Delivery of Final Clinical Study Report |
|
*** |
|
CONFIDENTIAL |
|
September 7, 2007 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #03
Appendix 4 — Budget
VIVUS, Inc.
Qnexa OB-303
Medpace Fee Estimate
***
|
CONFIDENTIAL |
|
September 7, 2007 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
***
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
Task Order #03
Appendix 5 — Payment Schedule
VIVUS, Inc.
Qnexa OB-303
Payment Schedule
As set forth in this Agreement, professional service fees totalling $*** will be paid by VIVUS for professional services rendered by Medpace according to the following schedule.
***
Pass-through expenses will be billed to VIVUS on a monthly basis as incurred. The first Wire-Transfer payment will be invoiced prior to any site becoming active to ensure funds are available for site payments. Medpace will pay sites immediately following receipt of Wire-Transfers. Medpace will invoice VIVUS on a monthly basis and payments will be made by VIVUS within seven days of invoice receipt.
Payment Information and General Conditions
***
Inflation
The fees stipulated in the fee estimate include inflation for the duration of the study as specified in this proposal. Any significant shift in timelines will require a revision to the fees.
|
CONFIDENTIAL |
|
September 7, 2007 |
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.
|
Appendix 6 |
Transfer of Obligations Form |
CONFIDENTIAL
Directions: Complete a form for each clinical study where Sponsor obligations have been transferred in accordance with 21 CFR Part 312, Subpart D (Responsibilities of Sponsors). Forward the completed form to Sponsor’s Regulatory Affairs Department for submission to the applicable regulatory agencies.
|
Drug: |
VI-0521 |
Study ID: |
OB 303 |
|
Study Title: |
A phase III randomized, double blind, placebo controlled multicenter study to determine the safety and efficacy of VI-0521 in the treatment of obesity in adults with obesity-related comorbid conditions. | ||
|
CRO Name: |
Medpace, Inc. | ||
|
CRO Address: |
*** | ||
OBLIGATIONS TRANSFERRED TO MEDPACE: x THE APPROPRIATE BOX(ES).
o All obligations in 21 CFR 312, Subpart D (Responsibilities of Sponsors) have been transferred to Medpace.
x The following obligations have been transferred to Medpace:
Sec. 312.32: IND Safety Reports
x Promptly review safety information. *Sponsor will be notified within one (1) business day of discovery of significant new or serious adverse events or risks, or any unusual frequency of reactions with respect to the drug.
o Notify all participating investigators in a written IND safety report of any AE associated with the drug that is both serious and unexpected.
o Notify the FDA in a written IND safety report of any AE associated with the drug that is both serious and unexpected.
Sec. 312.53: Selecting investigators and monitors
x (a) Select qualified investigators
x (b) Control investigational drug shipment
x (c) Obtain information from investigators
x (1) Signed Form FDA-1572
x (2) CV or other qualification statement
x (3) Clinical protocol outline
x (4) Financial disclosure information
x (d) Select qualified monitors
Sec. 312.54: Emergency research
o (a) Monitor the progress of all studies involving an exception from informed consent.
o (b) Monitor such studies to identify when an IRB determines that it can’t approve the research.
Sec. 312.55: Informing investigators
x (a) Provide sites with the current Inv. Brochure.
x (b) Inform investigators of new observations on the drug, particularly with respect to AEs and safe use.
Sec. 312.56: Review of ongoing investigations
o (a) Monitor the progress of all IND studies.
x (b) Secure compliance from noncompliant investigators or discontinue drug shipments and end the investigator’s participation in the study.
o (c) Review and evaluate the safety and efficacy results as it is obtained from the investigator.
x (d) Discontinue use of the investigational drug if it is determined to present an unreasonable and significant risk to subjects, notify all IRBs and investigators, and assure the return or alternate disposition of the drug from the investigators.
Sec. 312.57: Record keeping and record retention
x (a) Maintain adequate records showing investigational drug receipt, shipment, or other disposition. *Master Drug Logs will include the name of the Investigator to whom the drug is shipped, the date, and the quantity and batch of each such shipment.
x (b) Maintain complete and accurate records showing any financial interests of the investigator subject to 21 CFR 54.
x (c) Retain the records and reports required by the regulations for 2 years after the marketing application is approved, or if not approved, until 2 years after investigational drug shipment is discontinued and FDA has been notified.
o (d) Retain reserve samples of any test article and reference standard identified and used in bioequivalence or bioavailability studies.
Sec. 312.58: Inspection of sponsor’s records and reports
x (a) Permit FDA personnel to have access to and copy and verify any records and reports related to the clinical investigation.
x (b) Permit DEA personnel to have access to and copy records related to the shipment, delivery, receipt and disposition of any investigational controlled substance. Assure adequate storage precautions are taken for investigational new drug substances listed in any schedule of the Controlled Substances Act.
Sec. 312.59: Disposition of unused supply of investigational drug
x Assure the return (or alternate disposition) of all unused supplies of the investigational drug from each discontinued/terminated investigator; maintain written records of any disposition of the investigational drug.
Other
o Please describe any other applicable transfers below:
September 7, 2007
*** INDICATES MATERIAL THAT WAS OMITTED AND FOR WHICH CONFIDENTIAL TREATMENT WAS REQUESTED. ALL SUCH OMITTED MATERIAL WAS FILED SEPARATELY WITH THE SECURITIES AND EXCHANGE COMMISSION PURSUANT TO RULE 24b-2 PROMULGATED UNDER THE SECURITIES EXCHANGE ACT OF 1934, AS AMENDED.