MDA Venture Philanthropy Grant Contract Between Muscular Dystrophy Association, Inc. and Summit Corporation plc (for the development of DMD and/or BMD therapeutics)
| Confidential Materials omitted and filed separately with the Securities and Exchange Commission. Double asterisks denote omissions. |
Exhibit 10.2 |
MDA Venture Philanthropy Grant Contract
Between Muscular Dystrophy Association, Inc. and
Summit Corporation plc
(for the development of DMD and/or BMD therapeutics)
This MDA Venture Philanthropy Corporate Grant Contract (“Contract”) is entered into as of December 15, 2011 (the “Effective Date”), between Muscular Dystrophy Association, Inc., a New York not-for-profit corporation (“MDA”) and Summit Corporation plc, a company incorporated in England and listed on the Alternative Investment Market of the London Stock Exchange having its registered office at 00 Xxxxxx Xxxx, Xxxxxxxx, Xxxxxxxxxxx, XX00 0XX, Xxxxxxx (“GRANTEE”), to document the terms upon which MDA will provide funding for the Project (as defined below) and certain related matters. MDA and GRANTEE are sometimes referred to herein individually as a “Party” and collectively as the “Parties.”
Background
GRANTEE has identified certain small molecules which can transcriptionally upregulate the utrophin gene (such compounds and any other compounds having a similar mechanism of action and all any salts, esters, free acid forms, free base forms, solvates, polymorphs, stereoisomers, enantiomers, racemates, metabolites and prodrugs thereof to which GRANTEE has rights from time to time, including SMT C1100, being referred to herein as the “Project Compounds”).
The Parties believe that the Project Compounds may have utility in the treatment of Duchenne muscular dystrophy and or Xxxxxx muscular dystrophy (collectively, the “Disease”).
GRANTEE has submitted to MDA a grant application titled “SMT C1100 utrophin upregulator: Request for Phase I clinical trial funding,” a copy of which is attached hereto as Exhibit 1 (the “Grant Application”), for a grant for partial funding of its project to develop a new formulation for SMT C1100 to enable such compound to advance into a new Phase I clinical trial for the prevention, treatment or amelioration of the Disease (such project, as the same may be modified from time to time by the Steering Committee (as defined below) being referred to herein as the “Project”); and MDA desires to provide such funding on the terms and conditions set forth in this Contract.
GRANTEE has submitted to MDA a budget for the Project covering work through the receipt of the final report in analyzing the plasma exposure from dosing of the new formulation for SMT C1100 (the “Budget”), a copy of which is attached hereto as Exhibit 5.
It also is contemplated that GRANTEE will receive additional funding for the Project from Duchenne Partners Fund LLC, a Delaware limited liability company (“DPF”), pursuant to the Grant Agreement, dated on or about the date hereof, between DPF and Summit, as the same may be amended or modified from time to time (the “DPF-Summit Agreement”).
NOW, THEREFORE, in consideration of the foregoing and the representations and warranties and mutual covenants contained herein, the Parties, intending to be legally bound, agree as follows:
| 1. | Certain Definitions. |
The following terms shall have the following meanings as used in this Agreement:
“Affiliate” of a Person means any other Person which (directly or indirectly) is controlled by, controls or is under common control with such Person. For the purposes of this definition, the term “control” (including, with correlative meanings, the terms “controlled by” and “under common control with”) as used with respect to a Person, means the possession, directly or indirectly, of the power to direct, or cause the direction of, the management or policies of such Person, whether through the ownership of voting securities, by contract or otherwise.
“Control” and “Controlled by” means, with respect to any GRANTEE Technology, the possession by GRANTEE or its Affiliates of the ability to grant the right to access or use, or to grant a license or a sublicense to, such GRANTEE Technology as provided for herein without violating the terms of any agreement or other arrangement between GRANTEE (or any of its Affiliates) and any Third Party.
“Person” means any individual, firm, corporation, partnership, limited liability company, trust, business trust, joint venture, Governmental Authority, association or other entity.
“Third Party” means any Person other than GRANTEE and its Affiliates or MDA.
| 2. | MDA Translational Research Corporate Grant Policy Manual; Exhibits. |
| A. | In addition to the other terms and conditions contained in this Contract, funding for the Project by MDA is subject to the MDA Venture Philanthropy Grant Policy Manual, a copy of which is set forth thereto as Exhibit 3 (the “MVP Policy”), except for such provisions of the MVP Policy as are specifically made inapplicable to or are superseded by this Contract as provided herein. In the event of a conflict between this Contract and the MVP Policy, the terms and conditions specifically set forth herein will take precedence over the MVP Policy. |
| B. | This Contract includes the following Exhibits: |
| Exhibit 1: | Grant Application | |
| Exhibit 2: | Milestones and Funding Schedule | |
| Exhibit 3: | MDA Venture Philanthropy Grant Policy Manual | |
| Exhibit 4: | Steering Committee Confidentiality Agreement | |
| Exhibit 5: | Project Budget |
| 3. | Funding; Funding Schedule; Suspension and Modification of Funding; Use of Funds. |
| A. | Award of Grant. MDA agrees to award a research grant in the aggregate amount of Seven Hundred Fifty Thousand Dollars ($750,000) to GRANTEE for the Project (the “Grant”), which shall be issued in installments in accordance with the funding schedule set forth in Exhibit 2, subject to the funding conditions set forth in Section 3.B, the right of MDA to suspend funding as provided in Section 3.C and the right of MDA to modify the Grant and the payments under this Contract as provided in Section 3.D. |
| B. | Conditions to Funding. Each installment of the funding to be provided by MDA under the Grant award as contemplated by Section 3.A and Exhibit 2 is subject to, and conditioned upon, the satisfaction of the following conditions; provided that any such condition may be waived by MDA is its sole discretion: |
| (i) | the timely achievement (as determined in accordance with Section 4) of each Milestone (as defined in Section 4) scheduled to be achieved (as set forth in Exhibit 2) prior to the scheduled date for achievement of such Milestone; |
| (ii) | compliance by GRANTEE with the terms of this Contract in all material respects; |
| (iii) | the accuracy as of the date of this Contract and as of the date of the payment of such installment of the representations and warranties of GRANTEE set forth in Section 7; |
| (iv) | the delivery by GRANTEE to MDA of a certificate of a senior officer of GRANTEE certifying the satisfaction of the conditions set forth in clauses (ii) and (iii) above; |
| (v) | the delivery to MDA on or promptly following the Effective Date of documentation satisfactory to MDA establishing the availability of other financial resources that, together with the Grant and assuming that neither MDA nor DPF suspend or cancel the provision of any part of their grant funds to GRANTEE, are sufficient to complete the Project. |
| C. | Suspension of Funding. MDA reserves the right, in its sole discretion, to immediately suspend all funding under this Contract and terminate the Grant and this Contract if (i) GRANTEE fails to timely achieve any of the Milestones described in Section 4 and Exhibit 2 (unless any such Milestone is achieved by the applicable extended date, if any, agreed to by the Parties in writing as provided in Section 4), (ii) GRANTEE takes any of the actions specified in Section 6.A (unless, in the case of the replacement of the Principal Investigator, MDA has given its prior written consent to such replacement), (iii) any action, proceeding, or claim described in Section 6.B is commenced or asserted against GRANTEE, or (iv) GRANTEE breaches any of its material obligations under this Contract. |
| D. | Modification of the Grant and Award. GRANTEE acknowledges that payment of funds under this Contract by MDA is contingent upon the availability to MDA of sufficient research funds to fund the payments under this Contract and to fund MDA’s other research commitments. MDA retains right in its sole discretion, upon not less than [**] days’ prior written notice to GRANTEE, to revise the Grant and the amount and/or timing of payments provided for in this Contract based on the availability of funds and budgetary constraints. |
| E. | GRANTEE Financial Commitment. The Grant is subject to GRANTEE demonstrating the availability of other financial resources that, together with the Grant, are sufficient to satisfactorily complete the Project. In furtherance of the foregoing, GRANTEE shall provide to MDA during the term of this Contract all updates to the budget for the Project. GRANTEE also shall provide to MDA promptly (and in any event not later than [**] days) following the second fiscal quarter of each fiscal year of GRANTEE and not later than [**] days following the end of each fiscal year of GRANTEE) a statement of the out-of-pocket costs incurred by GRANTEE for the Project, a reconciliation of such costs against the budget for the Project, a statement of the costs that remain unpaid and the sources of funds that have been used to pay such costs and that are expected to be used to pay outstanding costs. |
| F. | Use of Grant Funds. GRANTEE shall use all Grant funds received from MDA solely for the Project. |
| G. | Effect of Suspension or Cancellation of Funding. MDA acknowledges that in the event of any decision by it or DPF to permanently suspend or cancel funding to GRANTEE under this Contract or under the DPF-Summit Agreement, GRANTEE may no longer have the financial resources necessary to complete the Project. A permanent suspension or cancellation of funding to GRANTEE under this Contract or under the DPF-Summit Agreement for reasons other than a breach of any such agreement by GRANTEE or a failure to satisfy the conditions for such funding set forth in the applicable agreement is referred to herein as a “Funding Suspension”. MDA agrees that (i) GRANTEE shall not have any liability to MDA solely as a result of the failure of GRANTEE to complete the Project solely as a result of a Funding Suspension and (ii) if GRANTEE permanently cancels or terminates the Project solely as the result of a Funding Suspension, MDA shall not be entitled to exercise its rights under Section 9 of this Contract. |
| 4. | Milestones. |
The milestones described in the attached Exhibit 2 (each a “Milestone”) represent pivotal evaluation points upon which further funding of the Project will depend. Both Parties agree to the timeline, Milestone descriptions, and (subject to the conditions to funding provided for in Section 3.B and the rights of MDA under Section 3.C and Section 3.D) payment schedule described in Exhibit 2; provided, however, any Milestone may be extended, in a written agreement signed by both Parties, for up to [**] months at the discretion of the Steering Committee. Milestone 3 will be considered to be timely achieved only if the Steering Committee determines by majority vote of the entire Steering Committee (as described in Section 5.B and 5.C) that such Milestone has been achieved by the date set forth in Exhibit 2. If the date for achievement of any Milestone is extended and such Milestone is achieved (as determined by the Steering Committee, in the case of Milestone 3) by such extended date, such Milestone shall be
deemed to have been timely achieved. MDA may, in its discretion, either provide or withhold funding during the extension period, but in the event such funding is provided, MDA will nonetheless retain its rights under Section A.XIV.2 of the MVP Policy and in Section 3.C of this Contract to cancel the Grant and terminate this Contract if the applicable Milestone, as extended, is not timely achieved.
| 5. | Steering Committee. |
| A. | The Parties shall establish a Steering Committee composed of one representative of GRANTEE, one representative of MDA, three outside experts (who are acceptable to both Parties) and for so long as DPF is entitled to appoint a representative to the Steering Committee pursuant to the DPF-Summit Agreement, one representative of DPF. If DPF ceases to be entitled to appoint a representative to the Steering Committee pursuant to the DPF-Summit Agreement, Summit shall immediately notify MDA and any representative or alternate appointed by DPF shall immediately cease to be a member of the Steering Committee. Each representative of Party shall be a full-time employee of such Party, unless the other Party consents in writing to the appointment of a representative who is not a full-time employee. Each representative or alternate appointed by DPF shall be a person with relevant scientific expertise who qualifies as an outside expert (as defined below) and to whom neither MDA nor GRANTEE has a reasonable objection. GRANTEE shall cause the DPF-Summit Agreement to include provisions relating to DPF representation on the Steering Committee that are consistent with this Section 5.A. As used herein, “outside expert” means a natural person having expertise pertaining to translational research who is not and has not been for the preceding three (3) years an officer, director or employee of or paid consultant to (i) either Party, (ii) any Affiliate of a Party, (iii) any beneficial owner of any Person that directly or indirectly owns or controls more than 5% of any class of securities of a Party or of any Affiliate of a Party and who does not otherwise have any direct or indirect financial or other interest in the Project or in any other business or competitive activity that could reasonably be expected to constitute a conflict of interest or to impair the ability of such person to exercise impartial judgment with respect to matters presented to the Steering Committee. Notwithstanding the foregoing, the prior or contemporaneous service by any person as a member of any steering committee, advisory committee or other committee having oversight or advisory responsibilities with respect to any other grant by MDA shall not make such person ineligible to serve as an “outside expert” under this Contract. Each Party also may designate an alternate for its representative on the Steering Committee who shall represent such Party and act in the place and stead of such Party’s representative on the Steering Committee if such representative is unavailable. Each such alternate shall meet the qualifications established above for a representative of a Party on the Steering Committee. Each Party shall designate its representative on the Steering Committee and any alternate by written notice to the other Party. Appointment of the outside experts on the Steering Committee will be confirmed by the Parties in writing. Each Party may, at any time upon written notice to the other Party, change its representative on the Steering |
| Committee or its alternate representative by written notice to the other Party. Any outside expert member of the Steering Committee may be replaced at any time upon the agreement of both Parties in writing. In addition, the Parties may designate, in writing, an alternate outside expert member. An alternate member may participate in meetings of the Steering Committee as a voting member in the absence of the applicable regular member. Outside expert members of the Steering Committee (including their alternates) shall be required to enter into written agreements that subject them to an obligation of confidentiality substantially equivalent to that provided for in Section 15 of this Contract, in substantially the form attached as Exhibit 4. The initial members of the Steering Committee are as follows: |
| MDA Representative: |
[**] Muscular Dystrophy Association, Inc. 0000 Xxxx Xxxxxxx Xxxxx Xxxxxx, XX 00000 | |
| MDA Alternate: |
[**] Muscular Dystrophy Association, Inc. 0000 Xxxx Xxxxxxx Xxxxx Xxxxxx, XX 00000 | |
| GRANTEE Representative: |
Xxx Xxxxxxx, PhD Senior Director, R&D [**] Summit Corporation plc 00 Xxxxxx Xxxx Xxxxxxxx, XX00 0XX, XX | |
| GRANTEE Alternate: |
Xxxxxxx Xxxxxx, DPhil Chief Scientific Officer [**] Summit Corporation plc 00 Xxxxxx Xxxx Xxxxxxxx, XX00 0XX, XX | |
| DPF Representative: |
Xxxxx Xxxx Preclinical Development Advisor [**] | |
| DPF Alternate: |
Xxxx Xxxxxxxx Business Advisor [**] | |
| Outside Expert: |
Xxxx XxXxxx President, PharMac LLC [**] | |
| Outside Expert: |
Xxx Xxxxxxx Independent Consultant [**] | |
| Outside Expert: |
Xxxx Xxxxxxxx Washington School of Medicine [**] | |
| B. | The Steering Committee shall determine (by majority vote of the entire Steering Committee) whether and when Milestone 3 has been achieved and shall meet as requested by the Parties to provide such advice and recommendations as the Parties may request from time to time and, if requested by either Party, shall determine whether any Interruption is the result of Scientific Failure in accordance with Section 9. The Steering Committee also shall have the responsibility for preparing (with assistance from GRANTEE) a quarterly summary of all significant reportable items under Section 10 of this Contract that relate to the progress and status of the Project. MDA may share this report with members of MDA’s Venture Philanthropy Advisory Committee (“MVP Committee”), subject to the members of the MVP Committee complying with the confidentiality restrictions with respect thereto set forth in Section 15 of this Contract. Members of the Steering Committee shall be entitled to conduct site visits at locations where activities relating to the Project are conducted. |
| C. | If practical in light of the requirements and timetable of the Project (including any ongoing clinical trial), the Steering Committee shall meet (in person or by telephone or video conference call) to determine whether Milestone 3 has been achieved by the date specified in Exhibit 2 (as such date may be extended as provided in this Contract). If because of the exigencies of any ongoing clinical trial or otherwise it is impractical for the Steering Committee to meet for such purpose, the Steering Committee (or any members thereof who are unable to meet) may be polled and the determination by a majority of the members of the Steering Committee, whether by vote at a meeting or by written communication to both MDA and GRANTEE, that Milestone 3 has been achieved shall constitute the majority vote of the entire Steering Committee. The failure of one or more members of the Steering Committee to attend any such meeting or to respond to any poll of the Steering Committee shall not affect any determination made by a majority of the members of the Steering Committee. |
| D. | GRANTEE shall submit to all members of the Steering Committee and contemporaneously therewith to MDA documentation relating to Milestone 3 sufficient for the Steering Committee and MDA to evaluate and determine whether or not such Milestone has been achieved and shall provide such |
| information as soon as it is reasonably able to do so. MDA and GRANTEE shall cooperate to cause the Steering Committee to make such determination as soon as reasonably practicable following receipt of such documentation from GRANTEE; provided, however, that nothing in this Section 5 shall preclude any member of the Steering Committee from requiring additional information from GRANTEE in order to make such determination. |
| 6. | Notice of Certain Actions. |
| A. | GRANTEE agrees that it shall give MDA at least [**] days’ prior written notice before taking any of the following actions; provided, however, that (i) such advance notice shall not be required where it would breach a contractual obligation of GRANTEE, violate any applicable law or regulation, or (based on the good-faith opinion of outside counsel to GRANTEE) trigger public disclosure requirements and (ii) with respect to transactions described in paragraph (1) or (2) below or a merger or consolidation described in paragraph (3) below, such notice shall not be required prior to the earlier of the execution of a binding commitment (whether or not subject to conditions) to consummate such a transaction or the occurrence of such transaction): |
| (1) | Enter into or permit to occur any Change of Control Transaction (as defined below); |
| (2) | Sell all or substantially all of its assets except for sales in the ordinary and normal course of its business as now conducted; |
| (3) | Liquidate, dissolve, merge or consolidate, or commence any proceedings therefor; |
| (4) | Replace the person serving as the principal investigator of the Project (the “Principal Investigator”) for the Project; or |
| (5) | File a petition in voluntary bankruptcy or requesting reorganization under any provision of any bankruptcy, reorganization or insolvency law or consent to the filing of any petition against it under any such law. |
| B. | GRANTEE agrees that it shall, within [**] days after it obtains knowledge thereof, provide written notice to MDA of (1) any action, proceeding, or claim which has commenced or been asserted against it in which the amount involved is more than $250,000, or if such claim could have a material adverse effect on the Project, impair MDA’s reputation, or prevent GRANTEE from performing its obligations under this Contract or materially impair GRANTEE’s ability to perform such obligations, (2) the termination or reduction of any other grant or source of funds intended for use in funding the Project or (3) a Cash Infusion (as defined in Section 8.A. |
| C. | As used herein, “Change of Control Transaction” means with respect to GRANTEE: |
| (1) | the acquisition by any individual, entity or group (within the meaning of Section 13(d)(3) or 14(d)(2) of the Securities Exchange Act of 1934, as amended) (a “Specified Person”) of beneficial ownership (within the meaning of Rule 13d-3 promulgated under the Securities Exchange Act of 1934, as amended) of fifty percent (50%) or more of either (a) the then outstanding shares of common stock of GRANTEE or any Person that directly or indirectly controls GRANTEE (the “Outstanding Common Stock”) or (b) the combined voting power of the then outstanding voting securities of GRANTEE or any Person that directly or indirectly controls GRANTEE entitled to vote generally in the election of directors of GRANTEE or such controlling Person (the “Outstanding Voting Securities”); provided, however, that for the purposes of this subsection C(1), the following acquisitions of securities of GRANTEE or such controlling Person shall not constitute a Change of Control Transaction of GRANTEE: (a) any acquisition by GRANTEE or such controlling Person, (b) any acquisition by any employee benefit plan (or related trust) sponsored or maintained by GRANTEE or such controlling Person or any corporation controlled by such Party or controlling Person or (c) any acquisition by any corporation pursuant to a transaction which complies with clauses (a) and (b) of subsection C(2) of this definition; |
| (2) | the consummation by GRANTEE or any Person that directly or indirectly controls GRANTEE of any acquisition, merger or consolidation involving any Third Party (a “Business Combination Transaction”), unless immediately following such Business Combination Transaction, (a) the individuals and entities who were the beneficial owners, respectively, of the Outstanding Common Stock and Outstanding Voting Securities immediately prior to such Business Combination Transaction beneficially own, directly or indirectly, fifty percent (50%) or more of, respectively, the then outstanding shares of common stock and the combined voting power of the then outstanding voting securities entitled to vote generally in the election of directors, as the case may be, of the corporation or other entity resulting from such Business Combination Transaction (including a corporation which as a result of such transaction owns the then-outstanding securities of GRANTEE or such controlling Person or all or substantially all of GRANTEE’s or such controlling Person’s assets either directly or through one or more subsidiaries) in substantially the same proportions as their ownership, immediately prior to such Business Combination Transaction, of the Outstanding Common Stock and Outstanding Voting Securities, as the case may be and (b) fifty percent (50%) or more of the members of the board of directors of the corporation resulting from such Business Combination Transaction were members of the Board of Directors of GRANTEE or such controlling Person at the time of the execution of the initial agreement, or of the action of the Board of Directors of GRANTEE or such controlling Person, providing for such Business Combination Transaction; or |
| (3) | GRANTEE or any of its Affiliates sells or transfers to any Specified Person(s) in one or more related transactions properties or assets representing all or substantially all of GRANTEE’s or such controlling Person’s business to which this Agreement relates at the time of such sale or transfer; |
provided, however, that the issuance by GRANTEE or any Person that directly or indirectly controls GRANTEE of new shares for cash in accordance with the rules of the AIM market (operated by the London Stock Exchange) and the Financial Services and Markets Xxx 0000 made with the primary objective of raising additional working capital for GRANTEE or such controlling Person (a “Public Offering”) shall not be deemed a Change of Control Transaction.
| 7. | Representations, Warranties and Covenants. |
| A. | GRANTEE and MDA each hereby represents and warrants to the other that it has full corporate power and authority to enter into this Contract, and it is not subject to any obligation that would impair or conflict with its ability to perform fully its obligations under this Contract. |
| B. | GRANTEE hereby represent and warrants to, and agrees with, MDA as follows: |
| (1) | As of the Effective Date, GRANTEE holds certain issued patents and patent applications relating to the commercialization of C1100, including a patent application claiming a novel composition of matter. Exhibit 6 sets forth a true and complete list of GRANTEE’s patents and patent applications relating to C100. |
| (2) | Based on available information after reasonable investigation, GRANTEE has all other intellectual property rights necessary to perform the Project as it is proposed to be conducted according to Exhibit 1 and has the power, authority and legal right to grant the MDA License (as defined in Section 9). |
| (3) | Based on available information, GRANTEE does not believe that the performance of the Project by it would infringe on the intellectual property rights of any Third Party. |
| (4) | GRANTEE is a public limited company duly organized and existing under the laws of England, is duly qualified to transact business and is in good standing in all states in which such qualification is necessary. |
| (5) | The Project and GRANTEE’s business are and will be conducted in accordance with the highest ethical standards set forth in the Helsinki Declaration of the World Medical Assembly and in compliance with all applicable national (United States federal, United Kingdom, etc.), state, foreign and local laws and rules and regulations and all applicable policies and protocols of the European Medicines Agency or any successor agency thereto (collectively, the “EMA”), U.S. Food and Drug Administration or |
| any successor agency thereto (collectively, the “FDA”) and other drug regulatory agencies, Human Subjects Review Boards and Institutional Review Boards of all collaborating institutions, and any other bona fide regulatory agencies governing clinical testing on human or animal subjects. |
| (6) | On the Effective Date and assuming no suspension or cancellation by MDA or DPF of funding to GRANTEE under this Contract or under the DPF-Summit Agreement, GRANTEE has available to it financial resources that, together with the Grant, are sufficient to complete the Project. GRANTEE has disclosed to MDA all other grants and funding sources intended to provide funds to be used in funding the Project, and GRANTEE is in compliance in all material respects with the terms of any contract or grants related thereto, and there has been no termination or reduction of any such grant or source of funds intended for use in funding the Project or any failure to meet any milestone or funding condition provided for in any such contract or grant. |
| (7) | There is no action, proceeding, or claim which has commenced or been asserted against GRANTEE in which the amount involved is more than $250,000, or which could have a material adverse effect on the Project, impair MDA’s reputation, or prevent GRANTEE from performing its obligations under this Contract or materially impair GRANTEE’s ability to perform such obligations. |
| (8) | GRANTEE has provided to MDA true and complete copies of all agreements and documents evidencing the Third Party Obligations (as defined in Section 9.D). |
| (9) | GRANTEE has delivered to MDA a true and complete copy of each freedom to operate study or similar study prepared by or on behalf of GRANTEE with respect to the Project and the Project Compounds and the intellectual property related thereto (each a “Freedom to Operate Study”) and shall provide to MDA a true and complete copy of each additional Freedom to Operate Study or any update to any such study, promptly after the completion thereof. |
| (10) | The initial Principal Investigator is Xxx Xxxxxxx, Ph.D. |
| C. | GRANTEE agrees that it shall conduct the Project substantially in accordance with the Grant Application, as the same may be modified from time to time with the consent of the Steering Committee and MDA. |
| 8. | Payments by GRANTEE. |
GRANTEE and MDA agree to the following commercial terms with regard to the development and commercialization of pharmaceutical products containing any Project Compound (all such pharmaceutical products being referred to herein, collectively, as the “Product”):
| A. | Development Funding Milestone Payment. In the event that prior to the first commercial sale of any Product that has been approved for use in the Field, GRANTEE receives a Cash Infusion (as defined below) and GRANTEE has not permanently ceased its research and development efforts directed towards developing and obtaining Regulatory Approval of the Products, GRANTEE will make a lump sum payment to MDA of Seven Hundred Fifty Thousand Dollars ($750,000) promptly following (and in any event not later than [**] days after) the receipt of such Cash Infusion (such payment being referred to herein as the “Cash Infusion Milestone Payment”). As used herein, “Cash Infusion” means (a) the receipt by GRANTEE or any of its Affiliates of funds or payments in one or more transactions in an aggregate amount exceeding [**] Dollars ($[**]) regardless of the source thereof, it being understood and agreed that (1) without limiting the foregoing, such funding may include the proceeds of the sale or issuance of any shares of capital stock or other securities of GRANTEE or any of its Affiliates, milestone or other payments received pursuant to any joint venture, collaboration, joint development or similar agreement and (2) in the event of the merger or consolidation of GRANTEE or any of its Affiliates with another Person, the cash, deposits, money market instruments, marketable securities and similar financial instruments held by such other Person and its Affiliates shall be counted as part of such Cash Infusion and the value of any proceeds paid to stockholders of GRANTEE and its Affiliates or (b) the direct or indirect purchase by a Third Party in one or more transactions of fifty percent (50%) or more of the outstanding shares of GRANTEE or any of its Affiliates or a merger or consolidation of GRANTEE or any of its Affiliates with a Third Party in which the proceeds of such purchase or merger to the shareholders of GRANTEE or such Affiliate have a value exceeding [**] Dollars ($[**]). |
| B. | Commercial Milestone Payments. |
| (1) | GRANTEE will make a lump sum payment to MDA of [**] Dollars ($[**]) (less the amount if any previously paid in respect of the Cash Infusion Milestone) not later than [**] days following the receipt of Regulatory Approval (such payment being referred to herein as the “First Commercial Milestone Payment”). As used herein, “Regulatory Approval” means the approval of the applicable regulatory authority in the United States or in the European Union of the use and sale of any Project Compound or Product for the prevention, treatment or amelioration of the Disease, excluding any pricing and reimbursement approvals; and, without limiting the foregoing, “Regulatory Approval” shall include the approval of a New Drug Application by the FDA or the approval of a Marketing Authorization Application by the EMA with respect to any Project Compound or Product for the prevention, treatment or amelioration of the Disease. |
| (2) | If the Product has received Regulatory Approval in the United States or the European Union and worldwide cumulative gross sales of the Product by GRANTEE and its Affiliates and their licensees exceed the aggregate |
| sum of [**] Dollars ($[**]) GRANTEE shall pay MDA an additional payment of [**] Dollars ($[**]) within [**] days after the end of the GRANTEE fiscal quarter during which such worldwide cumulative gross sales level is achieved. |
The foregoing payments described in this subsection B are referred to herein collectively as the “Commercial Milestone Payments” and together with the Cash infusion Milestone Payment are referred to herein collectively as the “Milestone Payments.”
| C. | Quarterly Payments. In addition to the Cash Infusion Milestone Payment and the Commercial Milestone Payments provided for above if the Product has received Regulatory Approval in the United States or the European Union, GRANTEE shall pay to MDA on a quarterly basis within [**] days following the end of each fiscal quarter of GRANTEE an amount equal to [**] percent ([**]%) of the worldwide Net Sales (as defined below) of the Product during such fiscal quarter. The foregoing payments are referred to herein collectively as the “Quarterly Payments.” |
As used herein, “Net Sales” means with respect to the Product, the gross amount billed by GRANTEE and its Affiliates and licensees for sales of the Product to a Third Party, less:
| (i) | discounts (including cash discounts and quantity discounts), cash and non-cash coupons, retroactive price reductions, charge-back payments and rebates actually granted to managed care organizations or to federal, state and local governments, their agencies, and purchasers and reimbursers or to customers; |
| (ii) | credits or allowances actually granted upon claims, damaged goods, rejections or returns of the Product; and |
| (iii) | taxes or duties levied on, absorbed or otherwise imposed on sale of the Product, including value-added taxes, healthcare taxes or other governmental charges otherwise imposed upon the billed amount (to the extent not paid by the Third Party). |
If GRANTEE or its Affiliate or licensee enters into an agreement with a Third Party for the purchase of the Product that provides discounts or rebates on the Product that are conditioned on pricing terms or conditions for purchase of another product or products sold by GRANTEE or such Affiliate or licensee, then the discount or rebate on the Product under such agreement shall be determined, for purposes of determining Net Sales under this Contract based on the weighted average of discounts or rebates for the Product and such other product(s) sold under such agreement for the applicable accounting period.
| D. | Payments in the Event of an Assignment of Rights. In the event GRANTEE assigns its rights to any or all of the Project Compounds or experiences a Change |
| of Control Transaction or to all or any part of the Product, GRANTEE shall, at the election of MDA, (i) cause such assignee to assume GRANTEE’s obligations under this Agreement with respect to the assigned rights or (ii) pay to MDA not later than thirty (30) days following the consummation of such assignment or Change of Control Transaction an amount equal to the greater of (x) [**] percent ([**]%) of the fair market value of the assigned rights or (y) such amount as is necessary to provide MDA with an internal rate of return on the Grant payments made by MDA under this Contract (without regard to the Commercial Milestone Payments) of [**] percent ([**]%) (such payment being referred to herein as the “Assignment Payment”). |
| E. | Payment Following Termination. If MDA terminates this Contract before MDA pays any grant funds to GRANTEE, or if MDA fails to make the first payment of grant funds to GRANTEE under this Contract despite the achievement of the relevant Milestone, then this Contract shall be null and void, and GRANTEE shall neither be liable for any Commercial Milestone Payment or any Quarterly Payment or Assignment Payment or for the MDA License grant provided for in Section 9.D of this Contract. IF MDA terminates this Contract after MDA makes the first payment of grant funds to GRANTEE, but before MDA pays the entire grant of $750,000 to GRANTEE, or MDA fails to pay to GRANTEE the full aggregate amount of payments contemplated by Section 3.A of this Contract, then (i) the Cash Infusion Milestone Payment, the First Commercial Milestone Payment and any Assignment Payment, whichever is due first, shall be reduced to an amount equal to [**] times the Grant funds actually paid by MDA to GRANTEE, and (ii) MDA shall not be entitled to receive any other Milestone Payment, any Quarterly Payment or Assignment Payment. |
| F. | The Parties further agree that this Contract supersedes in its entirety the MDA “Patents and Licensing Policy” included as Exhibit 1 to the MVP Policy. |
| 9. | Consequence of an Interruption. |
| A. | If, after MDA pays GRANTEE all grant funds in accordance with this Contract, an Interruption occurs (other than as the sole result of a Funding Suspension (as defined in Section 3.G), (i) if such interruption occurs for reasons other than Scientific Failure (as defined below) or Funding Suspension, GRANTEE shall promptly refund to MDA all amounts paid to GRANTEE under this Contract together with interest at the rate of [**] percent ([**]%) and (ii) the MDA License provided for in Section 9 shall become effective. MDA acknowledges that GRANTEE’s business model is not to take Products beyond Phase I itself but instead to license the further development and commercialization of Products to a commercial partner. As used herein, an “Interruption” shall be deemed to have occurred in the event: |
| (1) | (a) | GRANTEE and its Affiliates cease for a period of more than [**]days reasonable research, development and commercialization efforts directed toward developing and obtaining Regulatory |
| Approval of, and commercializing, Products, including without limitation using diligent efforts to obtain a Third Party development and commercialization partner for the Product; or | ||||
| (b) | it GRANTEE has licensed to a Third Party commercialization partner the right to develop and/or commercialize the Product and has not retained a right of reversion with respect to such rights in the event such licensee ceases to devote reasonable efforts to the research, development and commercialization of the Product (a “Right of Reversion”), such licensee ceases for a period of more than [**] days reasonable research, development and commercialization efforts directed toward developing and obtaining Regulatory Approval of, and commercializing, Products or ceases to sell Products in the United States or European Union; or | |||
| (c) | in the event the right to develop and/or commercialize the Product reverts from such Third Party licensee to GRANTEE, whether pursuant to a Right of Reversion, termination of the relevant agreements or otherwise, and GRANTEE fails to: (i) use reasonable efforts directed toward developing and obtaining Regulatory Approval of, and commercializing, Products, including without limitation using diligent efforts to obtain a Third Party development and commercialization partner for the Product (as contemplated by Section 9.A.1(a) above) or (ii) within [**] after the date of such reversion enter into a further bona fide transaction with a Third Party pursuant to which it licenses to such Third Party the right to continue the development and/or commercialization of the Product; and | |||
| (2) | MDA delivers to GRANTEE written notice (an “Interruption Notice”) stating MDA’s belief that an Interruption has occurred; provided that an Interruption shall not be deemed to have occurred if GRANTEE re-commences reasonable development efforts within [**] days after receipt of such Interruption Notice and such reasonable development efforts continue uninterrupted for a period of not less than [**] days. |
For the purposes of this definition, reasonable development efforts shall require the annual expenditure of not less than [**] Dollars ($[**]) on such efforts.
As used herein, “Scientific Failure” means that:
| (i) | the Project Compounds demonstrate unacceptable levels of toxicity or other adverse characteristics that make products containing the Project Compounds inappropriate for administration to humans; or |
| (ii) | the primary end point of any clinical studies of C1100 are not met; or |
| (iii) | the inability of GRANTEE (or its affiliates, agents or contractors) to develop a manufacturing process that would permit production of commercial quantities of Product in accordance with pharmaceutical good manufacturing practices with the consequence that it is impractical to commercialize the Products. |
In the event of a disagreement between the Parties as to whether there has been a Scientific Failure, such disagreement shall be resolved by the Steering Committee, and the determination of the Steering Committee shall be final and binding on the Parties.
| B. | GRANTEE hereby grants to MDA, effective upon the occurrence of an Interruption, an exclusive, worldwide, perpetual, irrevocable and royalty-free license under the GRANTEE Technology, with the right to sublicense, to research, develop, manufacture, have manufactured, use, sell, offer to sell, import and export the Project Compounds and the Products for use in the Field. Such license is referred to herein as the “MDA License.” As used herein, the “Field” means the prevention, treatment or amelioration of the Disease. As used herein, the “GRANTEE Technology” means all Patents, data, technical information, know-how, inventions (whether or not patented or patentable), trade secrets, processes and methods Controlled by GRANTEE used or useful in connection with the research, development, manufacture, use, sale, offer for sale, import or export of the Project Compounds or Products in the Field. As used herein, “Patents” means (i) the domestic or foreign patents and patent applications issued by or filed with patent authorities anywhere in the world which claim the composition or any method of use of any Project Compound or Product or the process of making any Project Compound or Product (ii) any continuation, continuation-in-part, divisional, and continued-prosecution applications of any patent applications included in the foregoing clause (i), and (iii) any patents issuing from any patent applications included in the foregoing clauses (i) and (ii), including any renewals, extensions, patents of addition, supplemental protection certificates, revivals, re-examinations, and reissues thereof. |
| C. | Promptly following the written request of MDA following the effectiveness of the MDA License, GRANTEE shall (i) deliver to MDA or MDA’s designee the GRANTEE Know-How Package, (ii) assign (or cause to be assigned) to MDA or MDA’s designee all of GRANTEE’s right, title and interest in and to all regulatory filings and approvals, if any, owned by GRANTEE and its Affiliates relating to the Project Compounds and the Product in the Field, (iii) assign to MDA or MDA’s designee any contract rights owned by GRANTEE and its Affiliates relating to the research, development or commercialization of the Project Compounds and/or the Product in the Field and (iv) to the extent they have not been transferred, grant (or cause to be granted) to MDA or its designee cross-reference rights to any relevant drug master files relating to the Project Compounds and/or the Product in the Field. As used herein, the “GRANTEE Know-How Package” means (a) all data and study results conducted with respect to the Project Compounds, including all compound structure information, |
| toxicology studies, clinical trials, formulation studies and manufacturing and regulatory activities relevant to the exploitation of the MDA License, (b) standard operating procedures for assays relating to the Project Compounds, (c) all manufacturing process and procedures and (d) any physical stocks of the Project Compounds. Such GRANTEE Know-How Package may be delivered in electronic form with all necessary format information and documentation as may be necessary to enable MDA or its designee to effectively use the information contained in the GRANTEE Know-How Package. GRANTEE shall provide MDA or MDA’s designee with reasonable access to and reasonable assistance of GRANTEE’s personnel and contractors for the purpose of effectively transferring such technology to MDA or MDA’s designee. |
| D. | MDA acknowledges that (i) other granting organizations may receive from GRANTEE similar field-specific licenses to the same Project Compounds and Product in their respective therapeutic areas, (ii) in connection with the in-license or acquisition of certain rights included in the GRANTEE Technology, GRANTEE has entered into agreements, and may in the future enter into additional agreements, that oblige any licensee of a Project Compound, including MDA if the MDA License is in effect, to make royalty or other payments in connection with development and commercialization of the Project Compounds and/or the Product (the “Third Party Obligations”), and (iii) the MDA License is made contingent on MDA’s or MDA’s sublicensee’s explicit assumption of such aspects of the Third Party Obligations as are required to support any development or commercialization efforts MDA or its sublicensees may undertake following the effectiveness of the MDA License; provided, however, that if a sublicensee of MDA assumes such obligation with respect to the rights sublicensed by it, MDA shall automatically be released from such obligations. |
| E. | GRANTEE shall, at its sole option, file, prosecute, maintain or extend the patent applications included in the GRANTEE Technology (the “Licensed Patent Applications”). GRANTEE shall: (a) keep MDA and its sublicensees and assignees reasonably informed of any material developments or notices in connection with the prosecution of any Licensed Patent Application; (b) give MDA and its sublicensees and assignees a reasonable opportunity to comment on any GRANTEE’s response to such developments or notices that are intended to be filed in any patent office in relation to the Licensed Patent Applications and consider in good faith any such comments; and (c) give MDA and its sublicensees and assignees a reasonable opportunity to liaise and cooperate with GRANTEE in the prosecution and maintenance of the Licensed Patent Applications. In the event that GRANTEE elects not to continue to file, prosecute or maintain patent protection for any of the Licensed Patent Applications, MDA and or its sublicensee or assignee shall have the right, at its option, to maintain such Licensed Patent Applications on behalf of GRANTEE. Without limiting the generality of the foregoing, in no event shall GRANTEE provide MDA or such sublicensee or assignee with notice of abandonment of any Licensed Patent Application less than [**] days prior to its date of lapse. Neither MDA nor any sublicensee or assignee of MDA shall have any liability to GRANTEE for any failure to prosecute or maintain any Licensed Patent Application. |
| F. | GRANTEE shall have the right and responsibility to maintain the patents included in the GRANTEE Technology (the “Licensed Patents”). GRANTEE shall give notice to MDA of any desire to cease maintenance of any Licensed Patents, including payment of maintenance fees or other fees, as applicable, and, in such case, shall permit MDA or its sublicensee or assignee, to exercise its sole discretion to continue maintenance of such Licensed Patents at its own expense. Should GRANTEE elect to not maintain the Licensed Patents and MDA or its sublicensee or assignee elect to continue maintenance of the Licensed Patents, GRANTEE shall execute documents and perform such acts at GRANTEE’s expense as may be reasonably necessary to effect an assignment of any such patent rights to MDA or such sublicensee or assignee in a timely manner, so as to allow MDA or such sublicensee or assignee to continue maintaining such Licensed Patents. GRANTEE shall promptly give notice to MDA and any sublicensee or assignee of the grant, lapse, revocation, surrender, invalidation or abandonment of any Licensed Patent. Neither MDA nor any sublicensee or assignee of MDA shall have any liability to GRANTEE for any failure to maintain any Licensed Patent. |
| 10. | Reports. |
In addition to the reports required by the MVP Policy, GRANTEE agrees to contemporaneously (except where a reporting date is specified or applicable law prohibits) provide the following information to MDA, provided, however, that distribution of such reports or the information they contain within MDA will be limited to the extent reasonably possible to MDA’s Board of Directors, officers, and employees, members of the MVP Committee (provided that the distribution to the MVP Committee consists solely of the report contemplated by Section 5.B), and members of the Steering Committee with a need to know, and that such reports or the information they contain may not be shared with any other non-employees of MDA absent GRANTEE’s prior written consent:
| A. | Copies of GRANTEE’s regular investor reports and, to the extent available to prospective investors, all registration statements, prospectuses, preliminary prospectuses, and similar materials created by GRANTEE in connection with a prospective or actual Public Offering; |
| B. | To the extent not made simultaneously available to (or, in the case of verbal or non-written communications, made outside the presence of) a representative of MDA or a member of the Steering Committee other than the GRANTEE Representative, copies of all communications (including summaries of all verbal and other non-written communications) with the EMA, the FDA or other drug regulatory agency concerning issues that may substantially affect the Project, specifically including, without limitation, copies of any communications from the EMA, the FDA or other drug regulatory agency allowing the effectiveness of any Clinical Trials Application or Investigational New Drug Application or similar |
| application and all communications concerning adverse events associated with the use of the Project Compounds or Product that are both serious and unexpected (as those terms are defined in the applicable regulations of the EMA, the FDA or other drug regulatory agency) that may occur in relation to any clinical trial involving the Project Compounds or the Product; |
| C. | Copies of all Institutional Review Board communications about and approvals of any clinical trial relating to the Project Compounds or the Product; |
| D. | Copies of all documents confirming that research involving animals, whether conducted in-house or subcontracted, complies with all applicable federal laws and regulations; |
| E. | All final study reports relating to the Project; |
| F. | Copies of any published or unpublished papers resulting from an MDA research award, including papers intended for future publication whether or not the publication date is known (it being understood and agreed that journal embargoes will not be violated); |
| G. | The date of the first commercial sale of the Product anywhere in the world; |
| H. | Within [**] days after the end of each GRANTEE fiscal quarter, a report in a form reasonably satisfactory to MDA of the gross sales and Net Sales of the Product and such other reports in a form reasonably satisfactory to MDA as may be reasonably requested by MDA to determine the amounts payable to MDA under this Contract and showing the computation of such amounts; |
| I. | Within [**] days after the end of each GRANTEE fiscal quarter, a report of the amounts expended by GRANTEE with respect to the Project; and |
| J. | Summaries of all Third Party Obligations referred to in Section 9.D undertaken by GRANTEE. |
GRANTEE shall keep or cause to be kept such records as are required to determine, in a manner consistent with United Kingdom generally accepted accounting principles (“GAAP”) and this Contract, the expenditure of funds under this Contract and the sums payable to MDA under this Contract. At the request of MDA, GRANTEE shall permit MDA’s Vice President - Finance, MDA’s outside accountants (currently Ernst & Young LLP), and/or an independent certified public accountant or accounting firm appointed by MDA, at reasonable times and upon reasonable notice, to examine such records as may be necessary to determine, with respect to any period, the correctness or completeness of any report, expenditure of funds, or payment made under this Contract. MDA shall be responsible for the fees of such independent certified public accountants for the performance of any such audit, unless such audit discloses a variance to the detriment of MDA of more than [**] percent ([**]%) from the amount of the original report, budget, or payment calculation. In such case, GRANTEE shall reimburse MDA for such fees.
Following completion of the Project and up until such time as the Product is commercialized, GRANTEE shall submit annual progress reports to MDA on the preclinical and clinical progress made on the Product in the Field. Such progress reports shall describe in summary fashion GRANTEE’s efforts to bring the Product through EMA and FDA approval and to commercialization for the Disease and shall include a discussion of the actual number of staff and expenditures directed toward the commercialization effort since the preceding report. Progress reports shall also contain: the status of clinical development including a summary of any communications with regulatory authorities; a description of efforts to commercialize the Product by any sublicensee; and any other information that may be deemed pertinent to the commercialization effort.
| 11. | Indemnification. |
GRANTEE agrees to indemnify and hold MDA and its officers, directors, employees, volunteers, and agents harmless from any and all claims, lawsuits, liabilities, demands, damages, expenses, and losses (including, without limitation, attorneys’ fees and costs), including, without limitation, personal injury or death (each, a “Claim”), resulting from:
| A. | GRANTEE’s performance or failure to perform any of its obligations hereunder; |
| B. | Any and all research and other activities conducted by GRANTEE and any investigator or subcontractor of GRANTEE pursuant to this Contract and in connection with the Project, including, without limitation, clinical testing on human subjects; |
| C. | Any product developed in whole or in part from the Project, provided, however, that such indemnification obligation shall not apply to any Claim arising from an act or omission by MDA or its sublicensees under the MDA License on or after the effectiveness of the MDA License or an event that arises out of an act or omission of MDA or its sublicensees under the MDA License and occurs on or after the effectiveness of the MDA License; |
| D. | Any violation or alleged violation by GRANTEE or any investigator or subcontractor of GRANTEE of any applicable law, governmental rule or regulation, or policy or protocol in conducting the Project; |
| E. | Any infringement or alleged infringement of the intellectual property rights of a Third Party, provided, however, that such indemnification obligation shall not apply to any Claim arising from an act or omission by MDA or its sublicensees under the MDA License on or after the effectiveness of the MDA License or an event that arises out of an act or omission of MDA or its sublicensees under the MDA License and occurs on or after the effectiveness of the MDA License; |
| F. | Any untrue or alleged untrue statement of material fact contained in any registration statement, prospectus, preliminary prospectus, or other offer by GRANTEE to issue or sell securities, or any omission or alleged omission of a material fact required to be stated therein or necessary to make the statements therein not misleading, or any other violation or alleged violation of federal or state securities laws by GRANTEE. |
In each of the foregoing cases: (i) MDA shall notify GRANTEE of any Claim with reasonable promptness under the circumstances, (ii) if GRANTEE has acknowledged in writing to MDA its full responsibility for such Claim, GRANTEE may select counsel to direct the defense and settlement of such Claim, provided, however, that MDA may, at its election, participate in the defense of any Claim, and (iii) MDA shall reasonably cooperate with GRANTEE, at GRANTEE’s expense, in the investigation of, preparation of, and defense and settlement of any such Claim. GRANTEE shall promptly reimburse MDA for any indemnifiable losses, damages and expenses incurred by MDA or any other indemnified Person hereunder as and when such losses, damages and expenses are incurred, subject to submission of reasonable documentation of such amounts. In the event GRANTEE assumes the defense of any such Claim as provided above, GRANTEE may not settle any such Claim without the prior written consent of MDA.
| 12. | Insurance. |
GRANTEE hereby represents, warrants, and agrees that (i) it currently maintains general corporate liability insurance and clinical trials insurance with coverage limits of not less than [**] Dollars ($[**]), and (ii) during the term of this Contract and the duration of the Project and until all applicable statues of limitations have expired with respect to the activities conducted in connection with the Project, GRANTEE shall maintain coverage with at least the same coverage limits so long as it remains available on commercially reasonable terms. GRANTEE shall cause such coverage to name MDA as an additional insured and to contain a waiver of subrogation. GRANTEE shall deliver to MDA a certificate of insurance evidencing such coverage prior to the date the first grant payment is due and from time to time thereafter upon the request of MDA.
| 13. | Communications and Notices. |
Any notice or other communication required or desired to be given with respect to this Contract shall be in writing, and shall be deemed to have been validly served, given or delivered immediately upon delivery by courier or upon transmission by telex, telecopy, e-mail (with confirmation of receipt) or similar electronic medium to the Parties and their respective designated project information officers for this Project as follows, or to such other address as each Party designates to the other in writing:
| Muscular Dystrophy Association, Inc. 0000 Xxxx Xxxxxxx Xxxxx Xxxxxx Xxxxxxx 00000 | ||
| Attention: | [**] | |
| with a copy to: | [**] | |
| Summit Corporation plc 00 Xxxxxx Xxxx Xxxxxxxx, XX00 0XX, XX | ||
| Attention: | Xx Xxx Xxxxxxx, PhD | |
| Senior Director, R&D | ||
| [**] | ||
| with an e-mail copy to: | ||
| Xxxxxxx Xxxxxx DPhil | ||
| CSO | ||
| [**] | ||
| 14. | Termination. |
This Contract shall be deemed to be terminated upon the exercise by MDA of any right it may have to cancel the Grant, provided, however, that the following sections shall survive termination of this Contract: Section 5, Section 6, Section 8, Section 9, Section 10, Section 11, this Section 12, Section 13, and Section 15.
| 15. | Confidentiality, Publicity, and Publication. |
| A. | Confidentiality. As used in this Contract, the term “Confidential Information” shall mean all information provided by one party (the “Discloser”) to the other party (the “Recipient”) pursuant to this Contract that is either explicitly designated as confidential by the Discloser, or is of such a nature that the Recipient either knew, or should reasonably have known, that it was disclosed with an expectation of confidentiality. Confidential Information shall not include (i) information previously known to the Recipient, as evidenced by written records, (ii) information which becomes publicly available other than through a breach of this Contract, (iii) information obtained by Recipient from a Third Party having no obligation of confidentiality to the Discloser, or (iv) information independently developed by Recipient, provided Recipient can demonstrate that the independent development was by employees or agents of Recipient who did not have access to Confidential Information. Recipient shall not disclose to any Third Party or use in any manner Confidential Information other than as explicitly permitted in this Contract for a period of [**] years from the date of disclosure by Discloser; provided, however, that (a) in the case of information explicitly designated as a trade secret, such information shall not be disclosed while it remains subject to trade secret protection, and (b) in the case of reports provided under Section 10 of this Contract containing information submitted to a government agency or institutional review board, such information shall not be disclosed until such time as it becomes available to the public pursuant to the federal Freedom of Information Act or similar state or local law. Disclosures required of Recipient by law or regulation shall not constitute a breach of this Contract, provided that Recipient minimizes the disclosure to the extent legally possible and (if legally permissible) promptly notifies Discloser of the disclosure requirement. Each party shall ensure that any of its employees or agents who require access to Confidential Information are subject to an obligation of confidentiality substantially equivalent to that provided for in this Contract. |
| B. | MDA Public Information. Notwithstanding the foregoing, MDA retains the right to make public the title and amount of the Project, as well as a brief lay summary reviewed and approved by GRANTEE. All other Project-related information, including the substance of the grant application, any resulting progress reports, or other written or oral communications is considered confidential and will not be released to other than MDA’s Board of Directors, officers, and employees, the MVP Committee (provided that the distribution to the MVP Committee consists solely of the report contemplated by Section 5.B), and the Steering Committee without specific, written permission from GRANTEE. GRANTEE shall designate one representative as the Project Information Officer to oversee and authorize release of Project-related information. |
| C. | Publicity. It is understood that MDA and GRANTEE will make every effort to coordinate publicity related to the Project as funded by MDA, and that MDA will respect confidentiality agreements and proprietary information about this work. GRANTEE shall cause all GRANTEE-generated press releases and journal publications relating to the Project to include a prominent reference to the support received from the Muscular Dystrophy Association and its seminal role in the Project. GRANTEE shall submit to MDA all GRANTEE-generated journal articles related to the Project as soon as they are accepted for publication, together with information about embargo dates. Press releases and other public media generated by MDA will acknowledge GRANTEE by name. Except for uses of a Party’s name provided for elsewhere in this Section, neither Party shall issue any press release or other information designated for public release that is related to the Project and uses the name of the other Party unless such press release or information has been approved in advance in writing by such other Party. Each Party shall use all reasonable efforts, subject to the AIM Rules or other public disclosure requirements, to provide the other Party with a reasonable time to review such press releases and information, and each Party shall endeavor to give its approval or the reasons for the denial of its approval as soon as reasonably practicable after receiving such press release or information. Without limiting the foregoing, GRANTEE acknowledges that MDA contemplates that it will make a public announcement contemporaneously with each public announcement by GRANTEE that relates to the Project, the Project Compounds or the Product. Accordingly, GRANTEE shall provide MDA with such advance notice of any such proposed announcement by GRANTEE as may be necessary for MDA to prepare its own announcement and for the review and approval of such MDA announcement by GRANTEE. If, prior to the expiration of [**] full business days following the receipt by MDA of GRANTEE’s draft announcement, MDA notifies GRANTEE that MDA will also make an announcement contemporaneously with the GRANTEE announcement, GRANTEE shall not make its own announcement with respect to such matters other than contemporaneously with MDA’s announcement unless MDA fails to provide GRANTEE with MDA’s proposed announcement prior to the expiration of such [**] business day period; provided that nothing in this sentence shall restrict GRANTEE from complying with the AIM Rules or other public disclosure requirements. |
The Parties further agree that this Contract supersedes in its entirety the “Translational Research Corporate Grant Communications and Confidentiality Policy” referenced in Part II(8) of Section A and Part III of Section C of the MVP Policy and included as Exhibit 2 to such policy.
| 16. | Miscellaneous Terms. |
| A. | This Contract may only be amended or modified by a written instrument signed by both Parties. |
| B. | This Contract shall not be assigned by either Party without the prior written consent of the other Party, provided, however, that (1) either Party may assign this Contract without consent to (i) any wholly-owned subsidiary or Affiliate under common control, or (ii) any successor to substantially all of the business to which this Contract relates, (2) MDA may assign to DPF or any organization that is a successor thereto any or all of its rights under this Contract, including any or all of its rights with respect to the MDA License, the Licensed Patents and Licensed Patent Applications and (3) MDA or any assignee of MDA may assign any or all of its rights under this Contract, including any or all of its rights with respect to the MDA License, the Licensed Patents and Licensed Patent Applications to any other Person for the further development and/or commercialization of Products. Any purported assignment not made in compliance with this Section shall be null and void. Upon any such assignment by MDA, MDA shall be automatically released from its obligations hereunder to the extent such obligations are assumed by such assignee; and, in the event of any such assignment by DPF (as MDA’s assignee), DPF shall be automatically released from its obligations hereunder to the extent such obligations are assumed by such assignee. |
| C. | This Contract, together with the Exhibits hereto, embodies the entire agreement between the Parties and supersedes all prior agreements and understandings, written and verbal, relating to the subject matter hereof. In the event that any provision of this Contract shall be invalid, illegal, or unenforceable, the validity, legality, and enforceability of the remaining provisions shall not in any way be affected or impaired thereby. The headings in this Contract are for reference purposes only, and shall not limit or otherwise affect the meaning hereof. This Contract shall be binding upon and inure to the benefit of the Parties and their respective successors and, to the extent permitted by Section 16.B, their respective assigns. |
| D. | This Contract will be governed by the laws of the State of New York. |
| E. | Any dispute, controversy, or claim arising out of or in connection with or relating to this Contract (each, a “Dispute”) shall be submitted to, and settled by, arbitration pursuant to the Commercial Arbitration Rules of the American Arbitration Association at a mutually-convenient venue to be determined by agreement of the parties (or, in the absence of such agreement, by the arbitrators). Any award rendered shall be final and conclusive upon the Parties. The expenses of the arbitration shall be borne equally by the Parties, provided that each Party |
| shall pay for and bear the cost of its own experts, evidence, and counsels fee’s, except that in the discretion of the arbitrator, any award may include the cost of a Party’s counsel. The requirement to submit Disputes to arbitration shall not prevent either Party from seeking injunctive relief from an appropriate court or other tribunal to preserve its rights while resolution of such Dispute is pending. In the event that the amount in dispute exceeds $[**] ($[**]), MDA may require such arbitration to be conducted before and resolved by a panel of three (3) neutral arbitrators appointed as follows. Each of MDA and GRANTEE shall appoint a neutral arbitrator (each a “Party Arbitrator”) and the two Party Arbitrators shall jointly appoint the third neutral arbitrator, who shall serve as the chairperson of the arbitration panel. If the two Party Arbitrators fail to agree on the third arbitrator within [**] days following the appointment of the Party Arbitrators, then the third arbitrator shall be appointed by the American Arbitration Association. |
| F. | The relationship between the Parties to this Contact is that of independent contractors and not agents of each other or joint venturers or partners. Each Party shall retain sole and exclusive control over its personnel and operations. |
| G. | This Contract may be executed in counterparts, each of which will be deemed an original, but which together will constitute one and the same instrument. |
| H. | Except in the event of fraud, willful misconduct or death or personal injury arising from negligence, no Party shall be liable to the other Party for any indirect, consequential, special or punitive damages. |
[Signature page follows.]
AGREED AND ACCEPTED:
| SUMMIT CORPORATION PLC | MUSCULAR DYSTROPHY ASSOCIATION, INC. | |||||||
| By: | /s/ Xxxxxxx Xxxxxx |
By: | /s/ Xxxxxxx Xxxx | |||||
| Name: | Xxxxxxx Xxxxxx | Name: | Xxxxxxx Xxxx, MD | |||||
| Title: | Director and CSO | Title: | Interim President | |||||
EXHIBIT 1
GRANT APPLICATION
![]()
SUMMIT PLC
SMT C1100 utrophin upregulator: Request for Phase I clinical trial funding
Xxx Xxxxxxx PhD
11th February 2011
Executive Summary Summit is proposing to fast track assessment of a second readily available formulation of SMT C1100 in healthy volunteers. It is our belief that this approach will give SMT C1100 the best opportunity of being able to demonstrate its true potential as a viable DMD therapy.
Summit has estimated that $[**] will be required to fund this plan of work and we are seeking the financial support of the MDA-VP to provide $[**] to assist in advancing SMT C1100 into a new Phase I trial using a more appropriate formulation. We are currently looking to identify additional partners to commit to the remaining balance required. In return, Summit will contribute its considerable scientific expertise to support SMT C1100’s development and eventual commercialisation as it remains our commitment to work towards producing a new medicine for DMD.
DMD remains a disease for which there is no cure. Summit understands it is a race against time for patients and families and that if the challenge of finding a cure, or even a life-enhancing treatment, is to be met, every available opportunity must be fully explored.
| CONFIDENTIAL | Page 1 |
![]()
Introduction
Duchenne muscular dystrophy (DMD) is a lethal X-linked recessive muscle wasting disease caused by mutations in the dystrophin gene. Affected boys are ambulatory until about 12 years of age. They used die in their late teens but many patients can live into their twenties with respiratory support. Many boys show at abnormal ECG in the late stages of the diseases and cardiomyopathy is a general feature of the disease. The milder form of the disease known as Xxxxxx muscular dystrophy (BMD) also characterized by cardiac defects even though BMD patients can be ambulant in the 50 and 60s. Thus any therapy for the disease would need not only to target skeletal but also cardiac muscle.
Currently there is no effective treatment for DMD. Various strategies to alleviate the symptoms such as steroid treatment, anti-inflammatory agents and growth hormone and myostatin inhibitors which promote increased muscle mass. More recently, genetic approaches have been tested in DMD patient trials. In particular, read through of stop codons have been attempted in the 10-15% of patients that have stop codons. Atularen entered a phase 2b trial which utilised a six minute walk distance test (6MWD) as the primary efficacy endpoint as the ability to walk further after treatment is considered a major improvement in the quality of life for these patients. Unfortunately after conclusion of the trial, no increase in the distance travelled using the 6MWD test was reported. Exon skipping using antisense molecules has shown some dystrophin increase in trials utilising direct injection into muscle. Early data on systemic delivery shows restoration of dystrophin to a variable degree in patients. Next generation trials are continuing with constructs which increase the efficiency of delivery of the antisense oligonucleotides. The efficacy of this approach was demonstrated in the dystrophin/utrophin knock out mouse where restoration of muscle function was demonstrated. The main exon skipping efforts are currently directed at skipping exon 51 which will target about 13% of patients. To treat more patients different antisense sequences will need to be developed to target other exons and the regulatory authorities may treat each of these new constructs as a new drug. The ideal scenario would be to develop multi-exon skipping but this may only be achieved using AAV delivery which also has problems.
We have taken an alternative pharmacological approach to DMD developing a strategy which should be appropriate to treat all patients irrespective of their mutation and would also have the potential to target skeletal and cardiac muscle. Building on our work on the mdx mouse which demonstrated that the loss of dystrophin could be compensated for by increasing the levels of the dystrophin related protein, utrophin, we have been developing novel small molecules which can transcriptionally upregulate the utrophin gene. The demonstration that increased utrophin can prevent the muscular dystrophy in the mdx mouse has been confirmed by others. Our data from the mdx mouse suggested that increasing the levels of utrophin 2-3 fold would be of great benefit. We therefore screened for small molecules that would increase the levels of utrophin in the cell based assay 4-5 fold.
SMT C1100 was the final product of an exhaustive chemical screening and optimisation campaign. In this paper we present evidence demonstrating a significant reduction in dystrophic symptoms and increased muscle function in dystrophin deficient mdx mice. This was a comprehensive study looking at the beneficial effects of daily dosing of SMTC1100 on both sedentary and the more severely affected forced exercise model. If the results obtained using the SMT C1100 translated across to DMD patients then undoubtedly this would be a disease modifying therapy for DMD.
| CONFIDENTIAL | Page 2 |
![]()
Results
In vitro upregulation of utrophin
SMT C1100 was identified from an iterative analoging approach from initial hits identified using a human muscle specific utrophin-A promoter cell based assay (ref from review or add in figure). Mdx myoblasts were cloned from H-2K-tsA58 x mdx with an IFN / ts SV40 T Ag transgene in order to control proliferation and fusion. The screening line contains a stably integrated reporter consisting of 8.4kb of the human utrophin (UTRN) promoter linked to a luciferase reporter gene. The region of the utrophin promoter contained all the motifs known to control utrophin expression. This high throughput screening assay identified a number of luciferase inducing compounds that also have the ability to increase the transcription of the endogenous mouse UTRN, thus identifying compounds with both human and mouse activity eventually leading to the final optimized compound, SMTC1100.
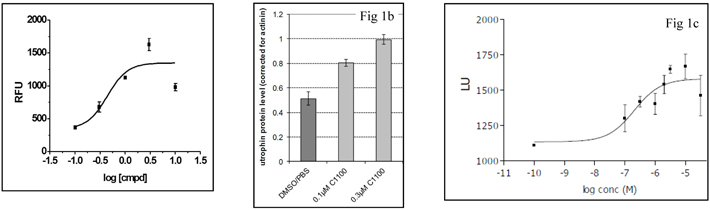
SMT C1100 shows a maximal increase of 5 fold compared to vehicle with an EC50 of 0.4µM (Fig 1a). In vitro dosing of human DMD muscle cells with SMT C1100 induces utrophin protein (Fig. 1b) when compared to vehicle dosing. Maximal utrophin protein increase of 100% above vehicle during the three days of dosing with 0.3µM was achieved. Human myotubes when dosed with SMT C1100 for 7 days demonstrated and increase of utrophin protein of around 50% when compared to vehicle with an EC50 of 0.2µM (Fig 1c).
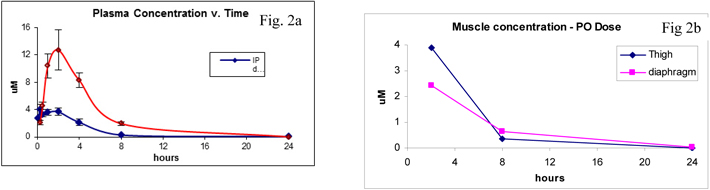
Plasma levels of SMT C1100
Single administration of 50mg/kg SMT C1100 by oral gavage demonstrates that levels of compounds are above the estimated efficacy level of 0.5µM in both plasma and muscle and are maintained for over 12 hours (Fig 2a and b respectively).
| CONFIDENTIAL | Page 3 |
![]()
In vivo upregulation of utrophin
To confirm the activity in vivo of SMT C1100, the dystrophin deficient mdx mouse was used to monitor any changes in the dystrophic phenotype after chronic dosing for several weeks. To confirm increases in utrophin expression after repeat daily dosing with SMT C1100, muscle samples were taken for RNA analysis and protein expression. Fig.3a demonstrates almost a two fold increase in utrophin mRNA as determined by quantitative PCR from mdx mice dosed daily with SMT C1100 for 28 days compared to vehicle. Muscle sections were also taken, Fig.3b demonstrates significant increases in utrophin protein quantified from western blots from heart, a muscle notoriously difficult to target with systemic administration of putative DMD therapeutics, and diaphragm, the skeletal muscle most affected in sedentary mdx mice. Fig. 3c

| illustrates a qualitative increase in sarcolemmal bound utrophin in the TA and EDL muscles after repeat dosing with SMT C1100. This data confirms SMT C1100 drives increased utrophin expression in vivo and more importantly demonstrates increased utrophin staining at the required site of action, the sarcolemma.
Benefits of daily dosing of sedentary mdx mice with SMT C1100
In the case of the sedentary mdx mouse, there is a significant triggering of muscle degeneration at around 4 weeks which continues for a further 4 weeks where limb muscles then appear to reach stasis and levels of regeneration remain stable. One muscle where continued development of necrosis is seen is the diaphragm muscle. The dosing schedule for SMT C1100 treated mice was a single daily administration from day 21 |
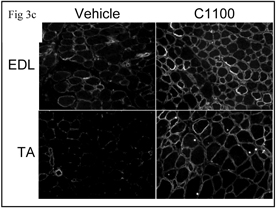
| |
| for a further 28 days. This period of dosing encompassed the necrotic degenerative phase resulting from dystrophin deficiency. | ||
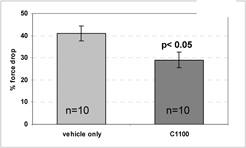
| CONFIDENTIAL | Page 4 |
![]()
| be able to function for longer. All of these endpoints are significantly improved in mdx dosed daily with SMT SMTC1100 for several weeks compared to mdx only dosed with vehicle. SMT C1100 addresses the primary cause of fibre loss by protecting the sarcolemma from damage as exemplified by resistance to eccentric contractions (Fig.4a) and a reduction in serum creatine xxxxxx xxxxxx (Xxx. 0x). At the point where the muscle necrosis is at a maximum, SMT C1100 reduces significantly the release of CK into the plasma (Fig4b, 15d after start of dosing) and when degeneration has stabilised there is still significant benefit seen as evidenced by continued lower levels of CK (Fig4b, 28d after start of dosing). This data also demonstrates that beneficial effects of SMT C1100 driven utrophin upregulation must occur within a few days after the start of dosing. |

|

| As the cycle of fibre degeneration – regeneration is being slowed by continued utrophin expression in SMT C1100 dosed mdx then the cytoplasmic signals to engage in muscle repair responses such as inflammation and fibrosis are reduced. In normal muscle this inflammatory protection response is dampened down as the proliferation of resident satellite cells fuse and reconnects the broken fibres. However, with the constant degeneration, these protection signals are not switched off, resulting in the continued influx of inflammatory cells and fibroblasts, leading to an increasing cascade of further fibre damage and loss of muscle “space” by fibrotic plaques. Treatment with SMT C1100 significantly reduces this damage by virtue of the reduced fibre regeneration. Blinded analysis by a board-certified veterinary pathologist of muscle sections from mdx mice dosed with either vehicle or SMT C1100 shows |
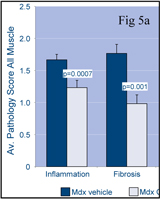
|
|
|
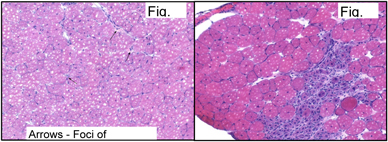
| CONFIDENTIAL | Page 5 |
![]()
a significant reduction in both inflammation and fibrosis. Whole muscle sections were rated with a pathology score on a scale from normal (0) – mild (1) – moderate (2) – severe. Results from the average of total scoring from the TA, EDL and soleus are shown (Fig. 5a). A qualitative example of the extent of inflammation from a SMTC1100 dosed EDL (Fig.5b) or vehicle dosed EDL (Fig. 5c) is shown where the SMTC1100 section was scored as mild (occasional mononuclear inflammatory cells in the inter-bundle connective tissue with focal aggregations of mononuclear inflammatory cells) and the mdx as moderate (multiple foci of mononuclear inflammatory cell infiltration in the inter-bundle connective tissue; occasional mononuclear inflammatory cells between individual muscle fibres). This data confirms the concept of reduced fibre damage due to utrophin localization leading to reduced inflammation and fibrosis.
Benefits of daily dosing of forced exercise mdx mice with SMT C1100
In order to increase the severity of the mdx muscular dystrophy, we used a forced exercise regime of chronic exercise, a strategy to worsen the murine pathology. Five week old mdx mice underwent forced treadmill exercise twice a week. The effects of daily SMT C1100 treatment was then evaluated on these forced exercise mdx mice.
| This exercise protocol significantly worsens in vivo parameters readily evaluated by non-invasive approaches, such as the grip and endurance tests. In particular, the exercise protocol induced the typical decrease of fore limb force in vivo over time, a reduction which is minimally observed in sedentary mdx. SMT C1100-treated mdx mice showed a significant protection against exercise- | ||
| induced fore limb weakness, in terms of maximal strength achieved and the increase in strength after 4 weeks of dosing (Fig 6a and 6b). After 4 weeks of dosing both values from the SMT C1100 dosed mdx were equivalent to those observed in wild type mice. |
| |
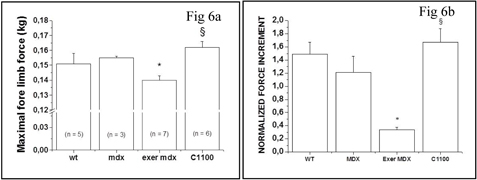

| CONFIDENTIAL | Page 6 |
![]()
effect was similar to that observed in exercised mdx mice treated with prednisolone, the standard of care for Duchenne patients. Significant synergy was observed when SMT C1100 was co-administered with prednisolone for five weeks, the forced exercise mdx were completely resistant to fatigue and were able to continue running as far as the sedentary mdx (Fig. 7) which equated to an increase in distance travelled of around 350% compared to the vehicle treated forced exercise mdx.
| Ex vivo analysis on isolated muscles from forced exercise mdx mice demonstrated that SMT C1100 exerted a significant amelioration of calcium-dependent functional parameters, which are typically modified in mdx muscles due to the altered calcium homeostasis | ||
| which in turn drives the rate of degeneration. In SMT C1100 treated EDL muscle fibres the strength-duration curve describing the mechanical threshold was significantly shifted toward the more positive membrane potential values and almost overlapped with that observed in wildtype myofibres (Fig. 8a). The rheobase value of SMT C1100 treated muscles approached the wildtype value (-69.3 ± 0.4 mV) which was about 5 mV less negative than that of non-treated exercised ones (-70.5 ±1.2 mV vs. -75 ± 1.5 mV: p < 0.05 |
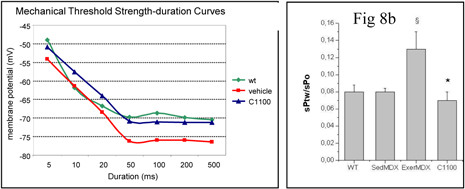
| |
| by Student’t test). Interestingly this parameter is not ameliorated by a partial increase in dystrophin expression by gentamicin treatment. Similarly, the ratio between twitch and tetanic tension was significantly reduced in SMT C1100 treated exercised mdx muscles with respect to untreated counterparts, again showing that SMT C1100 treatment generates similar values typically found in | ||
| wildtype EDL muscle (Fig 8b). The amelioration of calcium-dependent parameters was paralleled by a partial although significant 18% decrease of cytosolic Ca2+ level determined by fura-2 microspectrofluorimetry (data not shown), thus corroborating that the sarcolemmal bound utrophin from SMT C1100 can improve calcium-mediate mechanotransduction signalling. As one would predict from the reductions in cytoplasmic Ca2+ levels, there is a significant reduction in the necrosis seen in muscle sections with a corresponding increase in healthy fibres and reduction in fibres showing evidence of regeneration from the SMTC1100 foreced exercise mdx. Table 1 shows a 75% reduction in necrotic areas. |

| |
Footnote: The in vivo work described was commissioned by Summit and performed in the laboratories of Xxxx. Xxx Xxxxxx (Oxford, UK), Xxxx Xxxxxxxxx xx Xxxx (Bari, Italy) and Xxxxxxx Labs - West (Sacramento, US)
| CONFIDENTIAL | Page 7 |
![]()
Discussion
The data presented here illustrates the effectiveness of dosing a well-established model of DMD with a novel oral utrophin upregulator for several weeks. SMT C1100 induces increased levels of utrophin RNA and protein in human muscle cells and significantly reduces dystrophin deficient muscle pathology to such an extent that significant benefit on whole body strength and endurance is observed. Currently prednisolone and deflazacort are the only drugs approved by the regulators for DMD. We believe that fatigue testing of mdx after a regime of forced exercise is a good surrogate for the primary clinical endpoint which is used in DMD trials, i.e. the distance walked in 6 minutes (6MWD test). Dosing with SMT C1100 alone showed significant benefit in this surrogate model where the 50% increase in the distance walked would have achieved the required efficacy endpoint if translated over to the 6MD test in DMD trials. Unexpectedly dosing the combination of SMT C1100 and steroid for several weeks completely prevented fatigue of mdx mice undergoing the forced exercise regime. Thus, the combination of the two drugs with presumed different modes of action protect the muscle from fatiguing with exercise thereby allowing for significantly increased ambulation. High levels of long term steroid use have unwanted side effects however a steroid sparing therapy working synergistically with a utrophin upregulator has the potential to become the new standard of care for all DMD patients.
The great advantage of this approach is that it will be possible to treat all DMD and Xxxxxx patients, whatever their dystrophin mutation. In addition, it could also be used in combination with existing or other new strategies in the future including utrophin stabilisation strategies such as biglycan. In choosing a dosing route, an orally bioavailable product to be taken at home would be the ideal preference. In short SMT C1100 has the perfect profile, an oral drug suitable for treating all DMD patients.
In the recent Phase I clinical trial sponsored by BioMarin, SMT C1100 (BMN195) achieved plasma exposure marginally below the predicted efficacy levels. This is frequently a problem in Phase I trials which can often be circumvented using new formulations of the drug to increase bioavailability. From the data presented in this application only modest plasma levels of around [**] for several hours are needed to generate enough utrophin for substantial benefit and strongly supports the importance of retesting new formulations of SMT C1100 in new Phase I clinical trials to confirm that utrophin upregulation is a promising strategy for the therapy of DMD.
Lead up to Phase I Studies
[**].
BioMarin Phase I Study Results
| The trial utilised jet milled SMT C1100 suspended in [**]. This was a randomised, double-blind, placebo controlled single- and multiple-dose study in healthy male volunteers; ages 18-55. The single ascending dose level was a doubling dose from 25mg/kg up to 400mg/kg. | ||
| The most important observation was that there were no significant adverse events at the plasma concentrations achieved. This is very relevant as we believe that the levels achieved in the BioMarin trial are close to therapeutic levels of [**]. | ||
| As expected with a [**] formulation there was high variability within each group. |

| |
| CONFIDENTIAL | Page 8 |
![]()
This was seen in the minipig dosing and historically this is the major issue of [**] in man. However it is apparent from the error bars that some individuals did achieve plasma levels higher than the required efficacy level of [**] in the single doses. We believe the overall low exposure is undoubtedly due to inability of the body to emulsify the compound in such large volumes of [**]. Apparent loss of linear dose increase was not due directly to SMT C1100 bioavailability but more likely due to the change in the formulation (increase of API in [**]) which led to a further reduction in emulsification leading to reduced bioavailability.
| With no adverse side effects noted the study moved to ascending multiple doses for 14 days using 100, 200 and 400mg/kg dosing regimen. Once again no significant adverse events were noted even at the highest dose. Again high variability observed within each group with little difference in exposure between 100mg/kg and 400mg/kg groups. There was an apparent reduction in AUC and Cmax within the first week of repeat dosing which then remained constant for the remaining 7 days of dosing. Examining the data there is still a significant level of SMT C1100 within this constant level from around day 7 to day 14 which is estimated to be around 30% of required efficacy level. One explanation of this reduction is that the repeated insult of gastric irritation due to daily [**] administration could result in activation of gut CYPs and cause SMT C1100 metabolism issues resulting in lower absorption. |
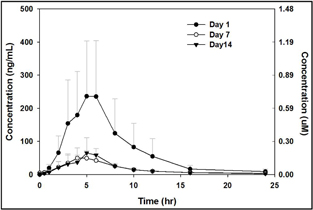
|
[**].
Conclusions from 1st Phase I Trial sponsored by BioMarin
Most importantly there were no reported significant adverse events and the compound was safe and well tolerated in all subjects including those who did achieve efficacy levels.
[**].
Summit’s conclusion from the data review and knowledge of the formulation is that the Phase I trial is inconclusive and does not lead to the conclusion that SMT C1100 development should be discontinued. The data points to the pressing need to repeat the Phase I with a more appropriate formulation.
Proposal to Test the Aqueous Microfluidised Formulation in a new Phase I
Summit believes the BioMarin results endorse the view that an alternative formulation could be more effective only needing to increase bioavailability in the order of a few fold to achieve and maintain significant efficacy levels.
[**].
Top Level Development Plan for SMTC1100
The current plan is to prioritise the aqueous microfluidised suspension formulation described above for use in a new Phase I trial in healthy volunteers.
[**].
| CONFIDENTIAL | Page 9 |
![]()
The planned Phase I trial is a double blind; placebo controlled ascending single oral dose, safety, tolerability and pharmacokinetic study. We have also included analysis of food effect, steroid combination, multiple daily dosing, and metabolism effects. Full study synopsis and plans will be made available if required. Time taken from the start of new non-GMP production of the new formulation for the PK cross over study in minipigs to the end of the Phase I is approximately 10 months.
The new trial will be deemed successful if the SMT C1100 plasma exposure using the new formulation is in excess of the efficacy level over the duration of a day after repeat dosing. This will confirm that there are no absorption or auto-induction issues with the compound and that dose levels will have been identified to translate into dosing regimens in Phase IIa DMD patient trials.
Total cost is approximately $[**].
Support Requested
Summit believes this proposal aligned with the research aims of the MDA-VP to identify therapies and a cure for DMD. We are therefore seeking financial support from the MDA-VP of $[**] to assist in advancing SMT C1100 into a new Phase I trial using a more appropriate formulation. We are currently looking to identify additional partners to commit to the remaining balance required. A successful outcome from this safety trial should enable clinical trials in DMD patients to start in 2012 and bring the first potentially disease modifying treatment for all DMD boys, irrespective of their genetic mutation, a significant step closer.
[**].
Without the requested funding from the MDA-VP, we are unable to progress further with the programme and fulfilling our objective of commencing DMD patient trials for utrophin upregulation in 2012.
| CONFIDENTIAL | Page 10 |
Exhibit 2
Milestone Schedule
| Milestone |
Date | Amount | ||||
| Milestone 1: Notice of Award Signed |
[**] | |||||
| First Grant Payment |
$ | [**] | ||||
| 2011 Total Payments |
Total $ | [**] | ||||
| Milestone 2: [**]. |
[**] | |||||
| [**] |
[**] | $ | [**] | |||
| Milestone 3: [**] |
[**] | |||||
| [**] |
[**] | $ | [**] | |||
| 2012 Total Payments |
Total $ | [**] | ||||
| Final report |
[**] | |||||
| Total Grant Payments |
Total $ | 750,000 | ||||
EXHIBIT 3
MDA VENTURE PHILANTHROPY GRANT POLICY MANUAL
|
|
MDA Venture Philanthropy, Inc. 0000 X. Xxxxxxx Xxxxx, Xxxxxx, XX 00000-0000 Telephone (000) 000-0000 — Fax (000) 000-0000 xxx.xxxxx.xxx | |

MVP RESEARCH PROJECT POLICY
Projects awarded by MDA Venture Philanthropy, Inc. (“MVP”) are governed by the policy set forth herein.
MDA and MVP support research aimed at developing treatments for the muscular dystrophies and related diseases of the neuromuscular system. These are the muscular dystrophies (among which are Duchenne and Xxxxxx); motor neuron diseases (including ALS and SMA); the peripheral nerve disorders (CMT and Friedreich’s ataxia); inflammatory myopathies; disorders of the neuromuscular junction; metabolic diseases of muscle as well as other myopathies.
MDA Venture Philanthropy (MVP) is MDA’s drug development arm. MVP acts within MDA’s Translational Research Program, and has the mission of identifying and overcoming the inherent regulatory, cultural, financial and logistical barriers to bringing to market new therapeutic agents for neuromuscular disease.
MDA defines Translational Research as research that transforms scientific discoveries arising from the laboratory, clinic or population into new clinical tools and applications that reduce neuromuscular disease incidence, morbidity or mortality. This broadly includes intervention development and Phase I/II clinical trials.
Terms of this policy are subject to revision
or alteration at any time
1
SECTION A
I. TYPE AND PURPOSE OF MVP GRANTS
MVP provides a mechanism to fund for-profit companies or academic institutions engaged in pre-clinical or clinical therapy development for any neuromuscular disease(s) covered in MDA’s program. Proposed projects should be focused on translational aspects of therapy development (e.g., optimization, scale-up, manufacturing, toxicology testing, and phase I/II clinical trials), and will be awarded with the understanding that there is a reasonable expectation that a therapeutic product will be brought to market by the MVP recipient, either during the tenure of the project, or subsequent to the award. A collaborative effort between academic or institutional researchers and companies in an appropriate field is preferred, and expertise may be offered to the applicant in the form of a steering committee with members agreed upon by both parties. The project may last for 1-3 years, with award amounts generally not to exceed $2 million per year. This award is highly competitive.
| I. | II. AWARD REQUIREMENTS |
| 1. | Proposed MVP projects must move a potential therapeutic agent closer to the market, and have a plan to make it commercially available (directly or indirectly). |
| 2. | Recipient will provide annual reports of expenditures to MDA. |
| 3. | Recipient will provide records of appropriate institutional/federal regulatory requirements. |
| 4. | Company recipients will be expected to demonstrate a fiscal commitment to the project that equals or exceeds the amount awarded by MDA. |
| 5. | Modular funding may be contingent upon achievement of pre-determined milestones as described in the grant application. For larger projects, achievement of milestones will be evaluated by a steering committee composed of one member of the MDA research staff, one member of the recipient organization and three outside experts for larger projects. For smaller projects, achievement of milestones will be evaluated on the basis of progress reports supplied to MDA. |
| 6. | Recipient will abide by the MDA policy agreement for intellectual property (Exhibit 1), or a modified version of this agreement that is acceptable to all parties. |
| 7. | Recipient and company will agree on a fair return on investment for MDA based on the stage of the project, financial investment by MDA and risk taken. |
| 8. | Recipient will abide by the MDA Translational Research Corporate Grant Communications and Confidentiality Policy (Exhibit 2), or a modified version of this agreement that is acceptable to all parties. |
| 9. | Recipient and MDA must come to agreement on a contract acceptable to both parties |
III. APPLICATION PROCEDURE
APPLICATIONS FOR FUNDING ARE NOT PUBLICALLY AVAILABLE.
Applications are made available to qualified applicants only. An application may be submitted and accepted at MDA’s sole discretion and is based on the nature of the research proposed and the qualifications of the applicant. In order to apply for funding, a Contact Form must be completed and submitted online for review (see xxxx://xxxxx.xxx/xxxxxx/xxxxxxxxxxx.xxx).
IV. DEADLINE DATES
MVP projects are reviewed on a rolling basis, and there are no deadlines for applications.
V. APPLICATION REVIEW
To ensure support of meritorious neuromuscular disease research, applications are peer-reviewed to assess their scientific merit and to evaluate their relevance to MVP’s goals. MDA’s Board of Directors has the sole authority to award all grants. Projects are reviewed under confidentiality agreements by outside reviewers (Exhibit 3) and by MVP’s advisory committee.
MVP projects are reviewed at two levels, firstly scientific and secondarily from a business standpoint.
2
The main criteria for scientific approval of MVP projects are:
Therapy Development Potential: Rationale of treatment modality; quality of preclinical data; results of toxicology studies to date
Plans for Proposed Research: Appropriateness of plan for advancing therapy towards commercialization; suitability of experimental plan; appropriate subcontractors used to get quality results; access to necessary institutional facilities and infrastructure
Ability to meet Regulatory Requirements: Manufacturing plan; regulatory compliance plan [for all projects beyond optimization]
Quality of Applicant/Co-Applicant(s): General excellence; expertise in clinical research; experience bringing therapies to market; quality of collaborators
The main criteria for business approval of MVP projects are:
Track Record of the Applicant: Experience of management, track record of attracting additional financing, track record of advancing therapeutics towards commercialization
Financial Stability of the Company (if relevant): Demonstration that the company has at least sufficient capital to complete the project with MDA investment, company has the ability to attract more funding, company has the necessary intellectual property to bring the therapy to market
Budget: Budget, including subcontracts, is reasonable for the proposed work
VI. PATENT AND LICENSING POLICY INFORMATION
Projects are subject to the Association’s Patent and Licensing Policy (Exhibit 1). By accepting a research project offered through MDA’s Research Program, the MVP recipient, all personnel contributing to and working on the project, as well as the Company/Institution with which they are affiliated, agree to be bound by the terms and conditions of MDA’s most recent policy on patents and licensing as described in Exhibit 1, or amendments of that policy described in the contract.
SECTION B
I. ELIGIBILITY FOR MVP RESEARCH PROJECTS
A corporate applicant must:
1. Be a biotechnology, pharmaceutical, or other related business.
2. Employ a qualified Principal Investigator for the duration of the award and during the conduct of the proposed project (at least 50% effort; precludes full-time employment at another Company/Institution).
An academic applicant must:
| 1. | Be a professional or faculty member at an appropriate educational, medical or research institution and be qualified to conduct and supervise a program of original research; |
| 2. | Have access to institutional resources necessary to execute the proposed research plan; and |
| 3. | Hold a Doctor of Medicine, Doctor of Philosophy, Doctor of Science or equivalent degree. |
| II. | II. DURATION OF RESEARCH PROJECTS |
Awards are for up to three years. Payments are contingent on the availability of research funds, achievement of predetermined milestones satisfactory to MDA, and confirmation that appropriate Company/Institutional requirements for human subject protection and animal care are current and on file (if applicable).
III. DELAY IN ACTIVATION
The activation of an MVP project by the MVP recipient may not be delayed. A MVP recipient who is unable to begin his or her project on its designated start date must relinquish the award and reapply.
3
IV. SUBCONTRACTS
MVP includes support for approved subcontracts. It is expected that the MVP recipient will manage collaborations with outside companies or academic investigators through a subcontract mechanism, with the understanding that MDA’s Research Grants Policy allows the payment of no more than 10% overhead to research organizations.
In connection with a subcontract, the MVP recipient will administer and account for all expenses of collaborators. This includes indirect costs. Appropriate documentation related to regulations governing animal and human subject use will be required from each subcontracted entity where applicable. The MVP recipient submitting the core research project application will be responsible for assembling and submitting to MDA such documentation along with a copy of each subcontract agreement prior to funding.
| III. | V. RESEARCH PROJECT PAYMENT |
Checks are made payable to the recipient company or institution and are issued upon achievement of milestones agreed upon in the contract. The milestones are set up such that the upfront payments should support the project up until the next milestone. If that milestone is deemed a failure, it suggests that the project should not go forwards and there should be no further expenses. Therefore, MDA would not make subsequent payments (with the caveat that MDA or the steering committee can approve a change in direction if there is an obvious alternative route forwards to develop the drug – at which point, alternate milestones would be agreed upon).
The company or institutional financial officer should establish an account from which research expenses may be paid under the terms of the approved award. MDA has the right to withhold or cancel payments at any time for non-compliance of Policies or failure to meet milestones.
| IV. | VI. AUTHORIZED EXPENSES |
When MDA deems them justified by the research plan, the expenses identified below are permitted under the MDA policies:
| 1. | Salary support for other scientists, technicians, research assistants, post-doctoral fellows, and graduate students and fringe benefits at levels appropriate to the Company/Institution. |
| 2. | Equipment and supplies necessary to complete the project’s milestones. Unless otherwise stipulated at the time of the award, equipment purchased solely with MDA funds belongs to and is considered the property of the academic Principal Investigator or the company to whom the research project was awarded. Computer hardware (i.e., PC’s, printers, monitors, etc.) is limited to a maximum of $5000 per grant. |
| 3. | Travel expenses: |
a. Must be directly related to the implementation of the research and/or expressly and solely for the purpose of reporting the results of MDA-supported research at suitable scientific or medical meetings and/or associated with getting the required regulatory approvals necessary to achieve the goals of the project.
b. Are limited to $1,000 maximum per year. With MDA approval exceptions may be made for projects such as clinical trials where travel to remote sites is an integral part of the project plan.
| 4. | Costs associated with submission of IND and to get FDA approval for a trial |
| 5. | Vendor Subcontracts: |
a. May include but are not limited to toxicology/biodistribution studies, product manufacture, formulation, data management, quality assurance and regulatory compliance (i.e., consultant or Contract Research Organization [CRO])
b. Maximum allowable for regulatory compliance consultation or CRO is $75,000 annually
| 6. | Indirect costs not to exceed 10% of direct costs. |
| V. |
| VI. | VII. UNAUTHORIZED EXPENSES |
The following expenses are NOT permitted under the MDA research program:
| 1. | Salaries, travel and/or housing related to sabbatical leaves; |
| 2. | Salaries for secretarial and/or clerical staff; |
| 3. | Purchase or rental of office equipment; (e.g. office furniture, filing cabinets, and copy machines); |
4
| 4. | Expenses normally covered by the indirect cost of the Company or the Principal Investigator’s institution; |
| 5. | Fees for tuition, registration or other fees relating to academic studies; |
| 6. | Membership dues, subscriptions, books or journals; and/or |
| 7. | Expenses for or related to moving from one institution to another. |
| 8. | Subcontracts to vendors in which the MVP recipient holds a proprietary interest. |
VIII. SUPPORT FROM OTHER SOURCES
| 1. | ALTERNATE FUNDING |
An Applicant may not apply for, use or accept MDA funds to support the same budgetary aspects of a research project already supported either by another source. If funds for the project in question become available to the MVP recipient from other sources during the review or tenure of an MVP research project, the MVP recipient must so inform MDA’s Research Department in writing. MDA will then make a decision about the allocation of its research award.
| 2. | SUPPLEMENTAL FUNDING |
Financial support for related but clearly non-overlapping aspects of a project from separate funding sources is permitted under MVP grants. For example, a company that already has a Small Business Innovation Research (SBIR) grant from the National Institutes of Health may still apply to MVP for funding related to additional distinct aims of the same project, provided that there is no scientific or budgetary overlap. Such supplementary funding must be disclosed, fully, to MDA as part of the research proposal or at the time such funding is received, and may be included in the matching funds required by MDA.
IX. BUDGET REVISIONS
During the review of the application, MDA may approve an award contingent on a reduction in budget. The applicant may choose to respond in writing within two weeks if the suggested budgetary changes are not acceptable, justifying in detail the need for the requested funding levels or suggesting a compromise. MDA will, in turn, respond within two weeks either accepting the compromise, or denying the request. If the request is denied, the applicant may choose to accept the award originally offered by MDA, or refuse the award.
MDA requires the submission of a revised budget when the funding for the project awarded is different than that originally requested in the proposal. The revised budget must reallocate the amount awarded for items requested in the original budget - except for any items specifically described in the contract that both parties have agreed to delete from the budget. A revised budget must be submitted to MDA’s Research Department within two weeks of receipt of the signed contract or within two weeks of acceptance of MDA’s required budgetary modifications.
To reallocate funds totaling more than fifteen percent (15%) of the current annual budget, the recipient must submit a written request to MDA’s Research Department for authorization. Such requests must include the amount of the reallocation and a detailed justification. Requests for budget revisions may be submitted up to four (4) weeks prior to the termination date of an award. Reallocations are permitted only during the current funding year.
X. UNEXPENDED FUNDS
Expenditures may not be committed against a grant after its expiration date except when authorized in writing by MDA.
At the termination of the project, unexpended funds may, under exceptional circumstances, be used for a period of either three (3) or six (6) months beyond the expiration date (no cost extension). The recipient must request in writing such an extension of the use of MDA funds. The request must state the amount of unexpended funds, how those funds will be used during the extension period and provide a detailed justification for the extension satisfactory to MDA. Such a request must be made no later than four (4) weeks before the termination date of the award. The originally approved budget remains in effect throughout the extension period, inclusive of all category maximums.
5
XI. CHANGE IN STATUS
The continued use of research project funds following any major change in status of the Principal Investigator requires prior written authorization from MDA. As described below, such changes include but are not limited to prolonged absence or withdrawal from the project.
1. WITHDRAWAL OR ABSENCE FROM THE PROJECT
When a Principal Investigator withdraws from a projector will be absent for an extended period of time, the MVP research project may be transferred to a new Principal Investigator, or his/her grant will be terminated and all unexpended funds plus unexpended accrued interest, if any, must be returned to MDA, accompanied by a Report of Expenditures, within eight (8) weeks of the withdrawal from the project.
If a project is continued under a new Principal Investigator, the Principal Investigator must write to MDA requesting authorization for such a continuation at least eight (8) weeks before the effective date of withdrawal or start of the absence from the project. The following documentation must be provided:
| a. | Effective date - month/day/year - of the change in Principal Investigator; |
| b. | Updated progress report on the project; |
| c. | Name, address and curriculum vitae of the proposed new Principal Investigator. |
| d. | Signed letter from the investigator referred to in item “c” above confirming that he or she is familiar with all aspects of the project and accepts full responsibility for the conduct of the research during the absence of the Principal Investigator. The proposed new Principal Investigator must indicate to MDA his/her familiarity with the specific aims of the project and agree to accept responsibility for all scientific and administrative aspects of the grant and also provide a statement about the availability of equipment, personnel, etc., necessary to conduct the research. |
2. CANCELATION OF GRANT
If, for any reason, the recipient of a research project must relinquish the award, the MVP recipient should promptly so notify MDA in writing. The notification should state the effective date of cancellation of the project. Unexpended funds plus unexpended accrued interest, if any, must be returned to MDA accompanied by a final Report of Expenditures within eight (8) weeks of the cancellation date.
MDA reserves the right to cancel a research project if circumstances render the individual on whose behalf the award was made unfit, unqualified and/or unable to perform under the terms and conditions of this MVP Policy. Such circumstances include, but are not limited to, abandonment of the project, loss of license, conviction of a crime, or withdrawal of insurance or other material institutional protections.
3. CANCELATION OF GRANT BY MDA
MDA has the option of canceling an award at anytime with notice for any of the following reasons:
| a. | If within ninety (90) days from the scheduled funding start date or the established deadline date for receipt of required reports, MDA has not received the required supporting documentation, e.g. copy of IRB, FDA, IACUC approval letters; IND confirmation; copy of informed/consent form(s); progress report; or other documentation as defined by the MVP Policies. |
| b. | Availability of Association resources are limited to the extent that continuation of funding of research projects must necessarily be placed on temporary or indefinite hold. |
| c. | For any violation of the guidelines governing MDA’s research grants program as defined by the Association’s MVP Policy. |
| VII. | 4. OWNERSHIP OF DATA |
In the event that the project is discontinued by MDA either due to failure by the MVP recipient to meet terms described in this policy or due to a default by the MVP recipient, and in so much that no provisions satisfactory to MDA have been developed for its continuance, all data produced with MDA funding up to the time of the cancellation will become the sole property of MDA.
6
XII. CURRICULUM VITAE/BIOSKETCH
Curriculum vitae of all investigators, advisors, co-investigators and post-doctoral fellows who will be participating in the execution of the research project must be provided to MDA with the research project application. When a project is underway, MDA must be informed immediately in writing of any change in personnel participating in the project, the reason(s) for such a change, and be provided the curriculum vitae or biosketch of any additional or replacement personnel.
SECTION C
RESEARCH REPORTS AND PUBLICATIONS
| VIII. | I. REPORT OF EXPENDITURES |
At the conclusion of each funding year of the grant, the designated financial officer of the recipient company or institution when the grant is awarded must submit a Report of Expenditures to MDA detailing the use of the awarded funds.
| IX. | II. REPORT OF PROGRESS |
Where Progress reports are required, Progress reports must be submitted on completion of each milestone as detailed in the milestone schedule. A final report must be submitted no later than four (4) weeks following the grant termination date. Where a steering committee is used, a transcript of the steering committee call will stand in lieu of progress reports.
| X. | III. PUBLICATIONS AND NEWS RELEASES |
All MDA Translational Research Grants, including MVP projects, are subject at minimum to the terms of MDA’s Translational Research Grants Communications and Confidentiality Policy (Exhibit 2)
Funds to support MDA’s research program come primarily from donations from private citizens. It is essential to the maintenance and growth of MDA and its research program that these donors be kept fully informed of the research progress their contributions make possible. Individuals and families affected by the neuromuscular diseases covered under its programs must also be kept fully informed of research progress. For these purposes MDA often issues press releases on newsworthy research developments and produces various publications for the public that report research activities. Such a press release or report may be issued on the occasion of the publication of an article in a professional journal or a presentation at a scientific or medical meeting. It is understood that MDA and the recipient will make every effort to coordinate publicity related to work funded by MDA, and that MDA will respect confidentiality agreements and proprietary information about this work.
To avoid misinterpretation of research results or the raising of false hopes about a possible treatment or cure for diseases covered under MDA programs, the Association requires the cooperation of the MVP recipient in providing MDA with advance prepublication copies of all articles and abstracts reporting the results of MDA-supported research which MDA shall keep confidential. MDA also requires the cooperation of its MVP recipients in participating in interviews as MDA may deem necessary. This cooperation will enable MDA to prepare press releases or other reports MDA issues on the research it supports.
If the recipient chooses to publish the results of the project, MDA requires that every such publication (print or on-line) - whether in peer-reviewed journals, meeting abstract formats, or in review articles or similar publications - contain the following statement or its equivalent: “Supported by MDA.”
SECTION D
HUMAN SUBJECTS/TISSUES AND ANIMAL RESEARCH
It is the responsibility of the Company/Institution to ensure that no MDA funds will be released for research involving humans and/or animals until the required documentation described below is on file with the appropriate official at the Company/Institution as well as MDA.
7
| XI. | I. INSTITUTIONAL REVIEW BOARD APPROVAL |
When human subjects, tissues and/or materials are to be used in a research project, it is the responsibility of the MVP recipient to ensure that the company/institution has the following on file:
1. A complete copy of the research protocol approved by the Company/Institution’s Human Subjects Review Board and a copy of that Board’s current approval notice;
2. A copy of the patient informed consent form(s) to be used.
A copy of the Board’s current approval notice and a copy of the patient informed consent form must be submitted prior to initiation of the human subject research, and upon renewal.
Projects must be in compliance with all policies, rules and regulations governing clinical trials including those of the federal regulatory agencies, the respective university and Company/Institution and MDA (if applicable). MDA must be advised about any amendments to the original research protocol (including the participant consent form) occurring prior to the commencement of or during the course of the research project. MDA must also be informed of any adverse events reported to the IRB, or other changes that may affect the project as it progresses.
| XII. | II. FOOD AND DRUG ADMINISTRATION APPROVALS |
When experimental drugs and/or experimental medical devices are to be administered to patients, the materials required in the “Institutional Review board approval” section “D” of this document are necessary. In addition, it is the responsibility of the MVP recipient to ensure that the following is on file at the company/institution, and that copies are sent to MDA:
| 1. | A complete copy of the Investigational New Drug (IND) and/or Investigational Device Exemption (IDE) application approved by the Federal Food and Drug Administration (FDA) and a copy of the FDA’s approval notice; and |
| 2. | Copies of all correspondence during the application and award periods between the FDA and the MVP recipient pertaining to the experimental drug(s) and/or device study. |
| XIII. | III. PATIENT CHARGES |
MDA requires that patients participating in experimental drug and/or device studies not be charged directly for any research procedures included under the project’s approved protocol. Patients must be fully informed about their responsibility for ancillary costs relating to participation in a research project — travel, lodging, food, etc.
IV. ANIMAL RESEARCH
MDA investigators may use animals and animal tissues for research purposes only when reasonable and practical alternatives do not exist. When completion of the milestones absolutely requires the use of animals and/or animal tissues, a detailed justification must be included in the research grant application submitted to MDA. The justification shall include statements confirming that Company/Institutional guidelines:
1. Are at least as protective as those of the National Institutes of Health;
2. Conform to all applicable laws and regulations;
3. Meet prevailing community standards for responsible scientific research;
4. Apply throughout the project to ensure the humane treatment of all animals involved in the project.
SECTION E
XIV.
| XV. | CONFLICT OF INTEREST |
Any potential conflict of interest the MVP recipient(s) or collaborator(s) may have relating to the project must be revealed. Such conflict would include (but may not be limited to) having a proprietary interest that may be affected by the outcome of a research project. It is expected that MVP recipients will observe the highest ethical standards in the conduct of research.
8
Exhibit 1:
PATENTS AND LICENSING POLICY
OF MUSCULAR DYSTROPHY ASSOCIATION, INC.
All MDA grants are subject to MDA’s Policy on Patents and Licensing. By accepting an MDA award for a research project, the MVP recipient or other personnel contributing to and working on the Project, as well as the Institution(s) with which they are affiliated, agree to be bound by the terms and conditions of MDA’s Patents and Licensing Policy.
MUSCULAR DYSTROPHY ASSOCIATION, INC. (“MDA”) understands that patents and licensing agreements may be sought on inventions resulting from research by the grant recipient supported in whole or in part by funds furnished by MDA; that such inventions should be administered so that they are introduced into public use as soon as practicable; and that such result will be achieved through granting permission to patent and license such inventions. Accordingly, it adopts the following policy:
| 1. | An invention (hereinafter “MDA invention”) resulting from the support in whole or in part to the grant recipient of funds awarded by MDA shall be reported to MDA promptly in writing. |
| 2. | If the university or other research institution or an individual investigator(s) associated therewith (“Institution” or “Investigator”) which is the recipient of financial support for the work leading to the MDA invention, has an established patent and licensing policy and procedure for procuring and administering inventions which are known to and accepted by MDA, or has an agreement with another organization, including agencies or departments of the U.S. Government relating to the MDA invention due to joint support, MDA will defer to that policy or agreement on the following terms: |
| a. | With respect to any such invention, the Institution or Investigator shall have the right to file a patent application, and if the Institution or Investigator decides not to file a patent application MDA shall be notified thereof within a reasonable time and thereupon MDA shall have the right to file a patent application. On any Institution- or Investigator-filed or MDA-filed patent application and on any patent obtained thereof or thereon, MDA shall have a nonexclusive, nontransferable, irrevocable, paid-up license to practice or have practiced for and on its behalf any such invention and to grant sublicenses thereunder. The patent application(s) and patent(s) obtained thereon shall embrace the United States and all countries outside of the United States. The inventions hereinabove contemplated shall include those made by employees or agents of the Institution or Investigator and third parties under the control of the Institution or Investigator. |
| b. | The Institution or Investigator will notify MDA in writing of any decision not to continue the prosecution of a patent application, pay maintenance fees, or defend a reexamination or opposition proceeding on a patent, in any country, not less than thirty days before the expiration of response period required by the relevant patent office. The Institution or Investigator will convey to MDA, upon written request, title to any such patent application or patent. |
| c. | The Institution or Investigator will make the invention available for commercial licensing upon reasonable terms and conditions. |
| d. | From the monies, if any, received from licensing the invention, MDA and the Institution or Investigator and all other parties shall share on terms mutually agreed upon by the Institution or Investigator and MDA, such terms to be determined prior to any licensing or commercial exploitation of the invention with MDA’s share being at least equal to the percent of total funding that MDA has financially supported the specific research project through grants and awards. |
9
| e. | In the event that it obtains a patent, license arrangement or other commercial exploitation of an invention, the Institution or Investigator shall make periodic reports to MDA with respect to its utilization of the invention and account for any income received by it by reason of exploitation of the invention. |
| f. | The Institution or Investigator or its licensee will use its best efforts to make MDA inventions available for the public benefit within a reasonable period of time. In circumstances of unreasonable delay, MDA shall have the right to require assignment of the patent or the invention to it; cancellation of any outstanding exclusive licenses granted relating to the invention and particularly under said patent; and the granting of any such licenses to a party designated by MDA on a nonexclusive royalty-free basis or on terms that are reasonable in the circumstances. |
| 3. | If the Institution or Investigator has no patent or licensing policy and procedure for administering inventions, MDA shall have the right to determine the disposition of the invention rights in any such case. |
10
Exhibit 2:
Translational Research Grants Communications and Confidentiality Policy
By accepting an MDA award for a research project, the company MVP recipient or other personnel contributing to and working on the Project, as well as the Institution(s) or companies with which they collaborate, agree to be bound by the terms and conditions of MDA’s Translational Research Grant Communications and Confidentiality Policy as they relate to the project for which the grant has been awarded. MDA in turn agrees to abide by the same provisions described in this policy. Additional provisions may be negotiated during the period before the recipient company formally accepts the MDA award. MUSCULAR DYSTROPHY ASSOCIATION, INC. (“MDA”) understands that information is sensitive and its release can be harmful to certain entities, and that proprietary information needs to remain confidential and protected. In turn, the recipient company or institution recognizes that dissemination of information about MDA funded research projects is important to the Muscular Dystrophy Association and the families that it serves. Such information is critical to ensuring the continued financial support of the Association by the public. If the recipient company or institution has an established confidentiality agreement that is known to and accepted by MDA, MDA may choose to accept, in addition, those terms of that policy or agreement that do not violate or supersede those of MDA’s Translational Research Grant Communications and Confidentiality Policy.
Policy Terms:
| 1. | MDA retains the right to make public the title and amount of all funded projects, as well as a brief lay summary. All other project-related information, including the substance of the grant application, any resulting progress reports, or other written or oral communications is considered confidential and will not be released to other than MDA staff without specific, written permission from the recipient company or institution. Recipient companies/ institutions should designate one representative (project information officer) to oversee and authorize release of project-related information. |
| 2. | It is understood that MDA and the recipient company or institution will make every effort to coordinate publicity related to work funded by MDA, and that MDA will respect confidentiality agreements and proprietary information about this work. All press releases or other information designated for public release that is related to the awarded project, whether generated by MDA or the recipient company, must be reviewed and approved by both the recipient company and MDA. Company-generated press releases and journal publications should include a reference to the support received by the Muscular Dystrophy Association and its role in the research project. Conversely, press releases and other public media generated by MDA will acknowledge the recipient company or institution by name. |
| 3. | Any and all project-related outcomes, achievements must be reported to MDA as specified in the grant paperwork as part of milestone and progress reports. |
| 4. | Copies of any papers resulting from an MDA research award must be sent to the MDA Research Department upon acceptance for publication, even if the publication date is several months away, or if there is no publication date known. Journal embargoes will not be violated. |
| 5. | Failure to adhere to this policy will result in termination of project support. |
11
Exhibit 3:
REVIEWER CONFIDENTIALITY AGREEMENT
THIS CONFIDENTIALITY AGREEMENT (this “Agreement”) is made by (the “Recipient”).
1. The Recipient has been requested to review an application for funding by Muscular Dystrophy Association. (“MDA”).
2. The Recipient acknowledges that, as a reviewer, he/she will receive confidential information from MDA and from one or more individuals and/or entities applying to MDA for funding (an “Applicant”), and the Recipient agrees to enter into this Confidentiality Agreement as a condition to reviewing these applications.
3. As used in this Agreement, the term “Confidential Information” shall mean all information received by the Recipient pursuant to an Applicant’s contract or application that is either explicitly designated as confidential by an Applicant, or is of such a nature that the Recipient either knew, or should reasonably have known, that it was disclosed with an expectation of confidentiality. Confidential Information shall not include (i) information previously known to the Recipient, as evidenced by written records, (ii) information which becomes publicly available other than through a breach of this Agreement, (iii) information obtained by Recipient from a third party having no obligation of confidentiality to an Applicant, or (iv) information independently developed by Recipient, provided Recipient can demonstrate that the independent development was by employees or agents of Recipient who did not have access to Confidential Information. Recipient shall not disclose to any third party or use in any manner Confidential Information other than as explicitly permitted in this Agreement for a period of five (5) years from the date of disclosure by an Applicant; provided, however, that (a) in the case of information explicitly designated as a trade secret, such information shall not be disclosed while it remains subject to trade secret protection, and (b) in the case of reports containing information submitted to a government agency or institutional review board, such information shall not be disclosed until such time as it becomes available to the public pursuant to the federal Freedom of Information Act or similar state or local law. Disclosures required of Recipient by law or regulation shall not constitute a breach of this Agreement, provided that Recipient minimizes the disclosure to the extent legally possible and (if legally permissible) promptly notifies MDA of the disclosure requirement. Recipient shall ensure that any of his or her employees or agents who require access to Confidential Information are subject to an obligation of confidentiality substantially equivalent to that provided for in this Agreement.
4. Recipient expressly acknowledges that the disclosure of an Applicant’s Confidential Information by Recipient will cause immediate, substantial, and irreparable damage to MDA and the Applicant for which monetary damages would not be a sufficient remedy. If Recipient breaches this Agreement in any manner, MDA and/or the Applicant shall be entitled to specific performance of this Agreement, an injunction restraining the Recipient from all further acts of disclosure and/or unauthorized use of Confidential Information, or both. These equitable remedies are in addition to all other rights MDA and the Applicant may have at law or in equity.
5. Recipient shall not assign this Agreement, his or her obligations hereunder, or any Confidential Information without the prior written consent of MDA
6. This Agreement embodies the entire agreement of Recipient and supersedes all prior and contemporaneous agreements and understandings, written and verbal, relating to the subject matter hereof. In the event that any provision of this Agreement shall be invalid, illegal, or unenforceable, the validity, legality, and enforceability of the remaining provisions shall not in any way be affected or impaired thereby. No alteration, amendment, or modification of this Agreement shall be made except by an instrument in writing signed by Recipient and MDA.
| AGREED AND ACCEPTED: |
| |||||||
| Tel.: |
| |||||||
|
|
Fax: |
| ||||||
| [Signature of Recipient] | E-mail: |
| ||||||
|
|
Date: |
| ||||||
| [Printed Name of Recipient] | ||||||||
| Address: |
|
|||||||
|
|
||||||||
12
EXHIBIT 4
FORM OF STEERING COMMITTEE CONFIDENTIALITY AGREEMENT
2
EXHIBIT 5
PROJECT BUDGET
Confidential Materials omitted and filed separately with the Securities and Exchange Commission. A total of one page is omitted. [**]
3
EXHIBIT 6
SUMMIT C1100 PATENTS AND PATENT APPLICATIONS
| REF |
TITLE (mnemonic) |
STATUS |
FAMILY |
APPLN NO. |
PUB/XXX NO |
PR DATE |
FILING DATA | |||||||
| [**] |
[**] | [**] | [**] | [**] | [**] | [**] | [**] |
Confidential Materials omitted and filed separately with the Securities and Exchange Commission. A total of one page was omitted. [**]

