CLINICAL TRIAL AGREEMENT
Exhibit 10.12
Rev. 16 November 2007
Page 1 of 17
THIS CLINICAL TRIAL AGREEMENT ("Agreement") is made by and between Xxxx Xxxxxxxxxx Cancer Research Center, a nonprofit corporation having a principal place of business at 0000 Xxxxxxxx Xxxxxx X., X0-000, Xxxxxxx, Xxxxxxxxxx 00000 ("Site") and Actinium Pharmaceuticals, Inc. with an address of 00X Xxxxxxx Xxxx, Xxxxxxx Xxxx, Xxx Xxxxxx 00000 ("Actinium"). This Agreement is entered into this July 19, 2012.
This Agreement provides for the conduct of a clinical investigation using a proprietary drug which is not, at this time, cleared for human use by the Food and Drug Administration. Any use of this drug must be pursuant to an Investigational New Drug Exemption issued to Actinium by the FDA. Aptiv Solutions, Inc., a Delaware corporation having a principal place of business at 0000 Xxxxx Xxxxxx Xxxxxx, Xxxxx 000, Xxxxxx, XX 00000 ("Aptly") has been engaged by Actinium to oversee and manage the Study.
In consideration of the mutual covenants and conditions set forth in this Agreement and for good and valuable consideration, the sufficiency of which is hereby acknowledged, the parties hereby agree as follows:
1. Definitions. When used in this Agreement, the listed terms shall have the following meanings:
|
a.
|
"Study" means the conduct of human research using the Study Drug manufactured by Actinium on Qualified Subjects at the Site pursuant to the protocol which has been reviewed and approved by the IRE and the FDA prior to the commencement of the Study.
|
|
|
b.
|
"Protocol" means the details of that certain clinical Study to be performed pursuant to this Agreement entitled A Phase I/II Study of Low Dose Cytarabine and Actinium-255 Hum195 in Older Patients with Untreated Acute Myeloid Leukemia. The Protocol, including any amendments is hereby incorporated by reference. and made part of this Agreement.
|
|
|
c.
|
"IRB" means the institutional review board of Site.
|
|
|
d.
|
"Study Drug" means the compound known as Actinium-255 Hum 195.
|
|
|
e.
|
"Study Data" means all of the data collected and records compiled during the Study relating to the conduct of the Study and/or the Study Drug. Study Data includes without limitation, all records prepared by the Investigator and all clinical research assistants, all Case Report Forms, all Screening records, all records on Qualified Subjects, Completed Subjects, Withdrawn Subjects, Uncompleted Subjects and Non-Qualified Subjects, all Informed Consent forms, all adverse/unexpected/serious or other reportable events, and all modifications, adjustments, suggestions for improvement of the Study Drug.
|
|
|
f.
|
"Potential Subject" is a patient or individual who could possibly participate in the Study.
|
|
|
g.
|
"Screening" is the process of identifying Potential Subjects and of conducting the examinations and tests necessary to select Qualified Subjects for the Study.
|
|
|
h.
|
"Qualified Subject" is a subject who, on inclusion in the treatment phase of the Study, has met all of the inclusion criteria and none of the exclusion criteria in the Protocol and has given his/her written Informed Consent to participate in the Study.
|
|
|
i.
|
"Completed Subject" is a Qualified Subject who has completed the Study and met the minimum attendance and compliance standards in the Protocol for evaluation of the safety and effectiveness of the Study Drug,
|
|
|
j.
|
"Withdrawn Subject" is a Qualified Subject who has been withdrawn from the Study because of treatment failure or adverse event, but who otherwise met the Protocol entry requirements.
|
Rev. 16 November 2007
Page 2 of 17
|
k.
|
"Uncompleted Subject" is a Qualified Subject who was initially included in the Study but who failed to complete the Study satisfactorily because of insufficient clinic attendance, poor compliance, voluntary withdrawal, and loss to follow-up or other Protocol violations.
|
|
|
l.
|
"Non-Qualified Subject" is a subject who has not met the inclusion criteria as defined in the Protocol.
|
|
|
m.
|
"Informed Consent Form" means the written form agreed upon by Actiniumand the Site in conformance with all applicable FDA regulations and guidances, and approved by the IRB for use in this Study.
|
|
|
n.
|
"Case Report Form (CRF)" means the report in the Actinuim format which is completed by the Investigator or his/her authorized designee documenting the use of the Study Drug in subjects.
|
|
|
o.
|
"FDA" means the Food and Drug Administration of the United States Department of Health and Human Services, and any successor government agency.
|
|
|
P.
|
"Investigator's Brochure" is a document describing the Study Drug, which is provided to the Investigator prior to the start of the Study.
|
|
|
q.
|
"Investigator" means Xxxx Xxxxx, M.D., Ph.D.
|
2. Compliance with laws and procedures.
|
a.
|
All parties shall conduct the Study in accordance with all applicable laws, regulations and guidances, as each of the foregoing may be amended from time to time. Without limiting the foregoing, the parties expressly agree to comply with 21 CFR 312 - Investigational New Drug Application, 21 CFR 50 Protection of Human Subjects, and 21 CFR 56 — Institutional Review Boards.
|
|
|
b.
|
The Site agrees to comply with the terms of this Agreement and all IRB and FDA procedures and applicable decisions for the Study.
|
3. Scope of Study
|
a.
|
This Study is governed by this Agreement. All parties agree that no Study Drug shall be used on a subject until the 1RB and the FDA have both approved the Study. After the FDA and IRB approval, the Protocol may only be amended when: 1) there is written agreement between the Site, Actinium, and the Investigator to amend the Protocol, and 2) any and all such amendments have been reviewed and approved by the IRB and FDA. No Protocol amendments shall be implemented until receipt of the IRB and FDA written approval. Nothing in this paragraph shall limit the Investigator's ability to act under 21 CFR 312.50, 312.60, 312.62, 312.64 (Subpart D).
|
|
|
b.
|
The Site agrees to strictly comply with all IRB procedures and policies which govern the review, approval and conduct of this Study.
|
|
|
c.
|
The parties agree that Screening for Qualified Subjects shall begin within thirty (30) days of receipt of the following: 1) written approval of the Study and the Informed Consent by the IRB, 2) notification by Aptly or Actinium that the FDA has granted the Investigational New Drug Application for the Study Drug, and 3) completion of Initiation Visit of the Site by Aptiv and Actinium for Study participation. The goal of the Study is to enroll up to ten (10) Qualified Subjects per year. The Site agrees to use its best efforts to complete subject enrollment as soon as practical, after commencement of Screening at each dose group.
|
Rev. 16 November 2007
Page 3 of 17
4. Responsibilities of Actinium
|
a.
|
Actinium represents and warrants that it has the authority to enter into this Agreement on its own behalf.
|
|
|
b.
|
Actinium agrees to provide to the Site and the Investigator the information necessary to properly conduct the Study, including without limitation, the Protocol, the Investigator's Brochure and data of any prior investigations of the Study Drug. Actinium agrees to provide any new information related to the safety and efficacy of the Study Drug as such information becomes available during the course of the Study. Actinium advises the Site and the Investigator that the effectiveness and safety in humans of the Study Drug have not been fully investigated.
|
|
|
c.
|
Actinium shall provide, free of charge, the necessary quantity of the Study Drug. Actinium or Aptiv shall ship the Study Drug only to the Site.
|
|
|
d.
|
Aptly shall monitor the Study and shall require evidence that IRB review and approval are obtained.
|
|
|
e.
|
Actinium agrees that Institution, its affiliates and all Study team members shall have the sole authority over the clinical care of the Study subjects and nothing in this Agreement shall prevent Institution or Investigator from taking any action which is, in the reasonable medical judgment of the Study team members, in the Study subject's best interest. Any time Actinium or Aptiv becomes aware of a significant Study subject safety issue it will communicate such information to Institution. Actinium further agrees to promptly report to Institution the results of any monitoring reports that could affect the safety of Study participants, influence the conduct of the Study, alter the Institutional Review Board ("IRB") approval to continue the Study, and/or affect the willingness of Study Subjects to continue in the Study. During the Study and after its completion, Actinium shall promptly report to Institution and the Investigator any Study results that could directly affect the safety or medical care of Study Subjects.
|
5. Responsibilities of Site
|
a.
|
The Site warrants and represents that Investigator is an employee of the Site, and is sufficiently qualified by training and experience to conduct the Study using the Study Drug. A true and complete copy of the Investigator's current curriculum vitae is attached as Exhibit B and made part of this Agreement.
|
|
|
b.
|
The Site warrants that the Investigator has never been involved in any investigation or research at the Site which was terminated by the FDA, National Institutes of Health (1'.411-1) or any sponsor for non-compliance.
|
|
|
c.
|
The Site warrants and represents that Investigator has not been disbarred under Section 306 of the Federal Food, Drug and Cosmetic Act, or any other section of said act or its successor, and further, that the Investigator will not use in any capacity, the services of any individual or entity which has been so disbarred, in any aspect of this Study. The Site agrees to notify Actinium immediately if the Investigator or any individual or entity involved in this Study is the subject of a disbarment proceeding or becomes disbarred.
|
|
|
d.
|
In addition to and without limiting the obligations of Section 2a above, the Site agrees to conduct the Study in strict accordance with this Agreement, the Protocol, all associated documentation provided by Aptiv (e.g. CRF, CRF Completion guidelines, User Manuals, and Regulatory Binder documentation), applicable regulations, and all conditions of approval imposed by the reviewing IRB or FDA. The Site shall permit the use of the Study Drug only on Qualified Subjects under Investigator's personal supervision only for the purpose of the Study. The Site shall not supply the Study Drug to any other person or entity not authorized under FDA'regulation to receive it, nor to any person for any purpose other than the Study. The Site shall not modify or alter the Study Drug. The Site shall maintain proper control of all Study Drug inventory and return of unused quantities of Study Drug as required by regulation and directed by Actinium. The Site agrees that the Investigator will personally supervise or perform all testing of the Study Drug involving human subjects.
|
|
Rev. 16 November 2007
Page 4 of 17
|
|
e.
|
The Site agrees to maintain all records and make all reports as required by regulation, the Study, the IRB and this Agreement.
|
||
|
f.
|
The Site agrees to use best efforts, on a diligent and continuous basis, to recruit Qualified Subjects, to prepare true and accurate Case Report Forms, to make all required reports, to complete the Study within the time limits set forth in this Agreement, and to perform all long-term follow-up examinations, visits and data collection as required by the Study and/or regulation from time to time. In addition, all CRF and Study Data shall be submitted to Aptiv within ten (10) days of written request. This provision shall survive termination or expiration of this Agreement.
|
||
|
g.
|
The Site shall exclusively use the Informed Consent. The Site agrees that the IRB approved consent form must be provided to and acknowledged by Actinium prior to use. The Site agrees that the Investigator shall not conduct any screening procedures, enroll any Potential Subject nor use the Study Drug on any Potential Subject who has not given written consent by signing and dating the specified Informed Consent form. The Site agrees that investigator shall personally ensure that all the requirements for obtaining informed consent are met.
|
||
|
h.
|
The Site agrees that Investigator will follow good medical practice and exercise the customary standard of care practiced in his professional specialty.
|
||
|
i.
|
The Site ensures that Investigator will provide sufficient accurate financial disclosure information to allow Actinium or Aptly to submit a complete and accurate certification or disclosure statement as required under 21 CFR part 54, as it may be amended from time to time. Further, the Site agrees that the Investigator shall promptly update this financial disclosure information if any relevant changes occur during the course of the Study and for one (1) year following completion of the study. The Site also agrees that Investigator will update this financial disclosure information within ten (10) days of request by Actinium or Aptiv. The Site understands that this information shall be submitted in any marketing application involving the Study Drug. This provision shall survive termination or expiration of this Agreement.
|
||
|
j.
|
The Site agrees to provide sufficient resources to the IRB to enable the IRB to operate as required by law, regulation and its own procedures.
|
||
|
k.
|
During the Study, and subject to the terms of this Agreement, Site agrees to use reasonable efforts to cause the Investigator to conduct the Study pursuant to the Protocol and to provide to Investigator reasonable access to all Site facilities, staff and resources which the Investigator determines necessary or desirable to the conduct of the Study. Without limiting the foregoing, the Site agrees to make available a Study coordinator, qualified by training and experience and reasonably acceptable to Actinium and Aptiv, to manage all administrative functions of the Study, including but not limited to, meeting with Actinium and Aptly. All such Site facilities, staff and resources used in the Study are subject to the supervision of the Investigator.
|
||
|
1.
|
The Site agrees to provide the facilities necessary to the conduct of the Study, and to notify Actinium and Aptiv promptly, but no later than 1 business day after the discovery, of any failure of the Investigator, the Site or the IRB itself, to follow any of the established protocols for the Study.
|
||
|
m.
|
The Site agrees to allow Aptiv and Actinium reasonable access to the study site and to facilities and staff as reasonably.needed to conduct long-term follow-up of Study subjects, at Aptiv's expense. The Site will ensure that the Investigator will be available, during regular business hours, to meet with a study monitor to review the status of the Study and discuss any pending issues. Aptiv will provide no less than five (5) days advance notice of monitoring visits and will use all reasonable efforts to coordinate the scheduling of the visits with the Investigator and Study Coordinator.
|
||
Rev. 16 November 2007
Page 5 of 17
|
n.
|
The Site agrees to allow Actinium and Aptiv reasonable access to Study Data, including without limitation, patient records (subject to patient consent), and Case Report Forms, as necessary for completion of the Study, long-term follow-up, and compliance efforts, at Actinium's expense.
|
|
|
o.
|
The Site warrants and represents that it will not use in the Study, in any capacity whatsoever, whether as employee, consultant, contractor or agent, the services of any individual or entity who has been disbarred under Section 306 of the Federal Food, Drug and Cosmetic Act, or any other section of said act or its successor. The Site agrees to notify Aptiv and Actinium immediately if any individual or entity involved in this Study is the subject of a disbarment proceeding or becomes disbarred.
|
|
|
p.
|
In the event the Investigator becomes unable to complete the Protocol for any reason, Site will, to the extent possible, propose a substitute Investigator with qualifications and experience at least equal to or greater than those of the Investigator for Actinium's approval, which approval shall not be unreasonably withheld. In the event Actinium and Site agree upon a substitute Investigator, this Agreement shall continue in full force and effect. If Actinium and Site are unable to agree on a substitute Investigator, this Agreement may be terminated in accordance with the provisions of this Agreement.
|
|
|
q.
|
Subparagraphs 5J-5P shall survive termination or expiration of this Agreement
|
|
|
r.
|
The Site and Investigator agree to notify Aptiv and Actinium as soon as possible, but in no event later than twenty-four (24) hours after each occurrence of an adverse, serious or unexpected event, or any deviation in the Protocol permitted by 21 CFR 312.60(aX2). The Investigator shall complete all reports when and in the manner required by 21 CFR 312.62 and 312.64. The Investigator shall make all other reports as required by 21 CFR 312.62 and 312.64.
|
|
|
s.
|
The Site agrees to cooperate with any study monitor designated by Aptiv to monitor this Study. The Site agrees to cooperate with authorized FDA employees conducting an audit or inspection, in the manner required by 21 CFR 312.68. The Investigator shall notify Actinium within 24 hours of any request for an audit of the Study by the Site, the FDA or any other governmental agency. If any inspection occurs, the Site will provide Actinium and Aptiv with copies of all auditor (including FDA and IRB) materials, correspondence, statements, forms and records that are received by the Investigator or the Site. Actinium and Aptiv will shall assist the Investigator in responding to any FDA or IRB correspondence and promptly implementing any necessary corrective action. This provision shall survive termination or expiration of this Agreement.
|
|
|
t.
|
The Site warrants that its Investigator has made all disclosures required regarding conflict of interest in connection with this Study.
|
|
|
u.
|
The Site hereby assures Actinium that the Study will be reviewed and approved by its IRB before any Study Drug is tested on a human subject, and further, that said IRB is functioning in compliance with the applicable regulations and all times. The Site shall provide, upon request, evidence of IRB approvals related to this Study in a timely manner, but in no event less than ten (10) business days after receipt, filing or request from Actinium or Aptiv, whichever is the case. This provision shall survive termination or expiration of this Agreement. The Site warrants that the Site has not, except as has been previously disclosed to Actinium and Aptiv in writing, in the last five (5) years been involved in any study which was terminated by any IRB, the FDA, National Institutes of Health (N111) or any sponsor for non-compliance.
|
Rev. 16 November 2007
Page 6 of 17
6. Payment
|
a.
|
Actinium acknowledge that it is Site's policy that the results of the Study must be publishable and the each Completed Subject of the Study, as set forth in the Site Budget and Payment schedule, attached to and made part of this Agreement as Exhibit C. All payments are gross in US Dollars; all approved invoices are net thirty (30) days.
|
Payment of all sums due hereunder shall be made by check payable as follows:
Xxxx Xxxxxxxxxx Cancer Research Center
Accounts Receivable J6-330
0000 Xxxxxxxx Xxxxxx X.
Xxxxxxx, XX. 00000
|
b.
|
Any additional payments must be approved in advance by Actinium in writing.
|
|
|
c.
|
The Site agrees to be responsible for invoicing Actinium in accordance with the Budget and Payment Schedule (Exhibit C).
|
|
|
d.
|
Any equipment (except for the Study Drug) purchased by the Site as part of the Protocol shall be owned by the Site, shall be physically located at Site, and shall remain the property of Site following completion of the Study.
|
|
|
e.
|
In no event is Actinium required to make any payment for any costs incurred with respect to Non-Qualified Subjects entered into the treatment phase of the Study or for any Subject who has not given written Informed Consent to participate in the Study.
|
|
|
f.
|
Nothing contained herein shall be construed as requiring Site, the Investigator or any Site research staff to work on any project or process which is prohibited by law or by any international treaty to which the United States of America is a party, or which may be harmful or detrimental to public health, patient safety or good clinical care or which may be considered to be immoral. No payment is subject to submission of favorable clinical results or evaluations.
|
|
|
g.
|
Notwithstanding the foregoing, in no event shall any payment be made under this Agreement which is contrary to 42 USC 1320a-7b, as it or any successor law may be in effect from time to time. In accordance with the statute, in no event shall the Investigator or any member of his immediate family, receive any payment, royalty, form of compensation, or emuneration of any nature, sort or description, for any use of all or any portion of the Study Drug by any hospital, clinic or other Site where he works. In no event shall any request for reimbursement or payment under any private or public health insurance carrier be made which is contrary to law.
|
Rev. 16 November 2007
Page 7 of 17
7. Publications
| Actinium acknowledge that it is Site's policy that the results of the Study must be publishable and the Investigator and others employed by Site or who are engaged in the Study be permitted to present at symposia, national or regional professional meetings and to publish in journals, theses or dissertations or otherwise in their sole discretion, the methods and results of the Study. |
|
The parties recognize that because this is a multi-center Study, there is a need for a coordinated approach to any publication or public disclosure of the data or results of this Study. To that end, there will be no publication or public disclosure of such data or results by the Site or Investigator until a multi-center publication is submitted for publication or presentation by Actinium, or its designee. However, if no multi-site publication is submitted by Actinium or its designee within twelve (12) months of the completion of the Study from all sites, the Site and the Investigator shall be free to publish for non-commercial purposes the Study results from there Site as follows. If the Site or the Investigator wishes to publish or publicly disclose Study and data or results the Site will submit any proposed manuscript or publication to Actinium for comment at least thirty (30) days prior to its submission for publication or other disclosure. The Site will review and consider in good faith comments received from Actinium during such thirty (30) day period. If requested to do so by Actinium, Site agrees to remove confidential information provided by Actinium prior to submitting the manuscript or publication, excluding Study data or results. Actinium will make every reasonable attempt to notify the Site within said thirty (30) days of receipt of the proposed publication whether it is desirable to file a patent application on any inventions contained in the proposed publication. In the event Actinium decides to pursue patent protection, Actinium shall have the right to defer publication for an additional sixty (60) days to permit the filing of any desired patent application.
|
8. Confidential Information
|
a.
|
The parties acknowledge that as part of the scientific collaboration between Actinium, Aptiv and the Site in connection with the Study, Aptiv or Actinium may find it necessary to disclose certain confidential and proprietary information and trade secrets of Actinium and/or Aptiv. Such confidential and proprietary information includes, without limitation, the Protocol, all intellectual property contained in the Study Drug, the design and manufacturing processes utilized to produce and test the Study Drug, the identity of Actinium's suppliers, data concerning scientific discoveries made by Actinium and/or Aptiv; Actinium's manufacturing strategies and processes; Actinium's marketing plans; data from Actinium's evaluations in animals and humans; Actinium's strategy for or status of regulatory approval; or Actinium's forecasts of sales and sales data, and any other information which by its nature would be considered confidential (hereafter referred to collectively as "Actinium Confidential Information"). Such Actinium Confidential Information shall remain the confidential and proprietary property of Actinium and shall be disclosed to Site's employees, affiliates or agents on a "need to know" basis, and who are bound by similar obligations to protect the Actinium Confidential Information from unauthorized disclosure. |
|
b.
|
The Site may find it necessary to disclose certain confidential and proprietary information and trade secrets of Site to Aptly Such confidential and proprietary information includes, any data, records or other information disclosed to Aptiv, or its designee, (hereinafter collectively, "Site Confidential Information"). Such Site Confidential Information shall remain the confidential and proprietary property of Site and shall be disclosed to Aptiv's or its designees, employees, affiliates or agents on a "need to know" basis. |
Rev. 16 November 2007
Page 8 of 17
|
c.
|
Subject to the terms and conditions of the Agreement, each party hereby agrees that during the term of the Study Agreement and for a period of five (5) years thereafter, neither party shall (i) publicly divulge, disseminate, publish or otherwise disclose any of the other party's confidential information without prior written consent; (ii) limit access to each party's confidential information to those of the other party's, co-workers and staff who are involved in the Study and have a need for such confidential information in connection with the conduct of the Study, and (iii) cause the return to the other party, as the case may be, any and all documents, drawings, sketches, designs, products or samples containing confidential information, together with any copies thereof, promptly upon termination of this Agreement or upon the other party's request therefore, provided that such obligations undertaken by the said party shall remain in force for seven (7) years after completion of the Study with respect to the Chemical Manufacturing and Control Section, Toxicity Studies or Performance Studies.
|
|
d.
|
Notwithstanding the foregoing, the obligations of confidentiality and nondisclosure shall not apply to the following information:
|
|
(1)
|
Information that was in the public domain prior to the date of disclosure to the receiving party coming into possession thereof, or becomes part of the public domain by publication or otherwise through no fault or unauthorized act or omission on the part of the receiving party;
|
|
|
(2)
|
Information that is disclosed to the receiving party by a third party legally entitled to disclose such information, as demonstrated by competent evidence;
|
|
|
(2)
|
Information that is disclosed to the receiving party by a third party legally entitled to disclose such information, as demonstrated by competent evidence;
|
|
|
(3)
|
Information that was rightfully in the possession of or already known to the receiving party as demonstrated by prior written records;
|
|
|
(4)
|
Information that is independently developed by the receiving party without reference to any confidential information, as demonstrated by competent evidence; or
|
|
|
(5)
|
Information that is required to be disclosed to a government authority or by order of a court of competent jurisdiction, provided that (a) such disclosure is subject to all applicable governmental or judicial protection available for like material; (b) reasonable advance notice is given to the disclosing party and disclosing party is provided with an opportunity to comment on such proposed disclosure; and (c) the receiving party take all reasonable steps to limit the scope of such disclosure.
|
|
e.
|
Site, in accordance with its policies and procedures, may post the Protocol on its internal database (referred to as "FYI") and share the Protocol, or portions thereof, as necessary i) to comply with applicable laws and regulations; ii) for internal patient care billing audits with Site's affiliates, and iii) to provide information to third party payors as necessary, in connection with the processing or payment of a claim submitted in relation to a Study subject. Site shall also be allowed to post a synopsis of the Protocol on its recruitment website.
|
In addition, the parties agree that the Investigator may disclose the title of the Study on his curriculum vitae and grant application(s).
Rev. 16 November 2007
Page 9 of 17
9 Intellectual Property
|
The parties acknowledge that it is unlikely that an invention will result during the performance of the Study. Site, on behalf of itself and its employees, consultants and agents, hereby exclusively, perpetually and irrevocably, conveys to Actinium a fully-paid and royalty-free, all rights, title to and interest in, including without limitation, all patent rights, copyrights, know-how, and other intellectual property of any nature, sort or description, which is created, learned, reduced to practice, discovered, or made in the performance of this Agreement or is related to or useful in connection with, the Study Drug or the Study Protocol ("Intellectual Property"). The Site and the Investigator shall promptly disclose all said Intellectual Property to Aptiv. The Site also represents that it has obtained sufficient authority to make this grant from all individuals that Site makes available to perform this Agreement and to provide good and clear title to Actinium. Additionally, Site, on behalf of itself and all of its employees, contractors and agents, hereby grants Actinium the right to file, prosecute and defend any such patent applications, at Actinium's .'.own cost and expense. Site agrees to render all reasonable assistance to Actinium in the filing, prosecution and defense of any such patent application at Actinium's own cost and expense.
|
|
The parties further acknowledge and agree that Actinium is the owner or authorized licensee of the Study Drug. Neither Site nor Investigator shall obtain any license to make, have made, sell, distribute, rent, lease, or otherwise transfer or use the Study Drug or Actinium Confidential Information, or their derivatives. Actinium Confidential Information is licensed for use only on and in combination with the Study Drug, and may not be used on or with third party products without Aptiv's prior express written permission. This Agreement grants no implied rights.
|
|
Aptiv on behalf of Actinium, hereby grants to Site a non-exclusive royalty free license to the Intellectual Property for Site's own internal nonprofit research and education related purposes.
|
10. Indemnification
|
a.
|
The Site shall, to the extent authorized by applicable law, indemnify, defend and hold harmless Actinium and Aptly, their agents and employees (collectively the "Indemnitees") from any and all liabilities, claims, actions, or suits (collectively "Claims") resulting from the negligence or wrongful acts or omissions of the Institution, the Investigator, their agents or employees pertaining to the activities of this Study and/or this Agreement, provided, however, that:
|
|
(i)
|
the Institution shall not indemnify, defend and hold harmless the Indemnitees from Claims arising out of the negligence or wrongful acts or omissions of the Indemnitees;
|
|
|
(ii)
|
the Institution is promptly, and in any event within thirty (30) days after an Indemnitee's receipt of notice of any complaint, claim or injury relating to any loss subject to this indemnification, notified in writing of any such complaint, claim or injury;
|
|
|
(iii)
|
the Institution has sole control over the defense and settlement of any such claim or suit, including the right to select defense counsel and to direct the defense or settlement of any such claim or suit, provided that Institution shall not admit fault or liability on behalf of any Indemnitee in the defense and settlement of such claim or suit; and
|
|
|
(iv)
|
the Indemnitees reasonably cooperate with the Institution and its legal representatives in the investigation and defense of any claims or suits covered under this Section 12(b). In the event that a conflict arises in the context of such an investigation or defense, the Indemnitees shall have the right at their own expense to select and obtain representation by separate legal counsel.
|
Rev. 16 November 2007
Page 10 of 17
|
b.
|
Actinium Indemnification.
|
|
Actinium shall indemnify, defend and hold harmless the Site, its trustees, officers, medical and professional staff, affiliates, employees, and agents and their respective successors, heirs and assigns (collectively the "Site Indemnitees"), against any liability, damage, loss or expense (including reasonable attorneys' fees and expenses of litigation) incurred by or imposed upon the Site Indemnitees or any one of them in connection with any third party claims, suits, actions, demands or judgments due to any side effect, adverse reaction, illness or injury occurring to any Qualified Subject as a result of the Study Drug, performance of a Study test or procedure, use of any equipment or supplies supplied by Actinium, or complying with the Protocol or any instructions provided by Actinium or approved by Actinium.
Actinium's indemnification shall not apply to any liability, damage, loss or expense attributable to (i) the negligent activities, reckless misconduct or intentional misconduct of the Site Indemnitees; (ii) failure of the Site Indemnitees to provide the current standard of care for subjects, excepting use of the Study Drug; (iii) failure of the Site Indemnitees to adhere to the terms of the Protocol for the Study or follow all prior written instructions provided by Actinium or Aptly, (iv) actions of the Site Indemnitees in violation of applicable laws or regulations , or (v) material breach of this Agreement by the Site Indemnitees. In no event shall Actinium have any liability of any nature, sort or description attributable to the exclusions of this sub-paragraph,.
This obligation to indemnify is subject to the Site Indemnitees giving Actinium written notice within two (2) business days of any claim, suit or demand and full control of any defense and settlements of such claim, suit or demand. The Site Indemnitees will also notify Actinium promptly in the event any one of them becomes aware of any potential claim, or likelihood of any potential claim of indemnification rights under this Section. Site Indemnitees will cooperate fully, at Actinium's expense, in the defense or settlement of any claim or action.
This Paragraph shall survive expiration or termination of this Agreement.
|
|
c.
|
Study-Related Injury.
Subject to Section 10(b), above, Actinium agrees to pay for all reasonable costs incurred for the care and treatment of any illness or injury to a Subject resulting from his or her participation in the Study.
|
I I. Insurance
|
The Site and Actinium shall, at its sole cost and expense, procure and maintain commercial general liability insurance or equivalent self insurance in amounts not less than $2 million per incident and $5 million annual aggregate with respect to the Study.
|
12. Term and Termination
|
Unless earlier terminated in accordance with its terms, this Agreement shall commence on the date when it is signed by all parties, (the "Effective Date", and shall continue in full force and effect until two (2) years after the Study has been completed.
|
|
a.
|
This Agreement shall be terminated immediately in the event that: 1) the authorization and IND issued by the FDA is withdrawn, 2) the approval of the IRB is withdrawn; or 3) the Investigator has not included any Qualified Subjects in the Study in accordance with Paragraph 3.c. and after the Study has been open to enrollment for twenty-four (24) months. |
Rev. 16 November 2007
Page 11 of 17
|
b.
|
Except as otherwise provided in this section, any party may terminate this Agreement upon sixty (60) days prior written notice in the event of any material breach by another party of any material term or condition hereof; provided such breach is not cured within said sixty (60) day notice period.
|
|
|
c.
|
Any party may terminate or suspend this Study immediately for the safety of Subjects, pursuant to applicable regulations. In such case, the party terminating or suspending the study will provide prompt written notice to the other party.
|
|
|
d.
|
Any party may terminate this Agreement upon written notice immediately in the event a party engages in criminal, unprofessional or fraudulent conduct.
|
|
|
e.
|
Aptiv may terminate this Agreement upon sixty (60) days prior written notice in the event that: I) the Protocol is suspended by the2) the Principal Investigator is unable to complete the Study and a substitute Principal Investigator cannot be agreed upon, or 3) if circumstances reasonably beyond Site's control preclude the Site from continuing the Study, and such suspension of the Study exceeds sixty (60) consecutive days or ninety (90) days in the aggregate in any year during the term (or renewal) of this Agreement.
|
|
|
f.
|
Aptiv may terminate this Agreement at any time upon one (1) month prior written notice to the Site. In such case, Aptly will provide finding of expenses actually incurred under the Study prior to the date of said notice or prior to a patient's completion of the Study if said completion is in the best interest of the patient as reasonably determined by the Investigator. Site may terminate this Agreement at any time upon ninety (90) days prior written notice to Aptiv.
|
|
|
g.
|
Any provision of this Agreement, which provides continuous enforcement or operation thereof after the termination hereof, shall survive the termination of this agreement.
|
13. Effect of Termination
|
Except as otherwise provided herein, termination of this Agreement shall not be construed to release either party from any obligation hereunder which has matured prior to the date of said termination. Upon termination of this Agreement, Site shall promptly return to Aptly the Study Drug, Study Data, including without limitation, all CRP and Actinium Confidential Information at Aptiv's expense.
|
14. Diversity in Study Population - Translation Services
|
The Site endorses the National Institute of Health's policy concerning the inclusion of minorities in study populations. The Investigator is encouraged to recruit patients into the Study without regard to ethnic background. The Site shall provide the consent form applicable to the Study in written form translated to the appropriate language to any non-English speaking minorities included in the Study. Any cost incurred by the Site for the development of a translated informed consent from will be agreed upon in advance and reimbursed by Aptly. The Site shall present Aptly with an invoice for translation services which Aptiv shall reimburse to the Site within thirty (30) days of receipt. The parties acknowledge that said translation costs are not included in the Study budget set out in Appendix B and are not included in the Total Cost of the Study.
|
Rev. 16 November 2007
Page 12 of 17
15. Communications
All medical/scientific and other communications, reports and notices shall be delivered by hand, by facsimile, by secure electronic means or sent by first class mail postage prepaid and addressed as follows:
|
If to Aptly:
If to Actinium:
|
Xxxxxxx Xxxxxxxx, Project Manager\
Aptiv Solutions 000 Xxxxxxxx Xx. Xxxxxxxxxxxx, XX. 00000
Xxxxxx Xxxxx, CEO
Actinium Pharmaceuticals, Inc.
000 Xxxxxxxxx Xxxxxx
Xxxxxx, XX 00000
|
If to Site : Xxxx Xxxxxxxxxx Cancer Research Center
Attn: Industry Relations and Clinical Research Support Xxxxxx
0000 Xxxxxxxx Xxxxxx X., X0-000
Xxxxxxx, XX 00000
If to Accounts Receivable: Xxxx Xxxxxxxxxx Cancer Research Center
Attn: Accounts Receivable
0000 Xxxxxxxx Xxxxxx X., .X0-000
Xxxxxxx, XX 00000
If to Investigator: Xxxx Xxxxxxxxxx Cancer Research Center
Attn: Xxxx Xxxxx, M.D., Ph.D.
0000 Xxxxxxxx Xxxxxx X., X0-000
Xxxxxxx, XX. 98109
In no event shall Site or the Investigator file any communication they are legally required to file with the FDA or any other regulatory agency, without first notifying Aptiv, unless otherwise requested by the FDA or other regulatory agency.
16. Use of Names
Except as otherwise required by law, each party agrees not to use or cite in any manner the name of the other party or its employees in any commercial or non-commercial advertising, article, press release or in any other forms of writing or publication medium without the prior written permission of the party or individual whose name or employee's name is to be used.
Rev, 16 November 2007
Page 13 of 17
17. General Provisions
|
a.
|
All rights and remedies hereunder are exclusive and not cumulative.
|
|
|
b.
|
This Agreement may be amended only by written agreement signed by all parties.
|
|
|
c.
|
It is expressly agreed by the parties hereto that the Site, the Investigator and Aptly are independent contractors and nothing in this Agreement is intended to create an employer relationship, joint venture, or partnerships between the parties. No party has the authority to bind any other.
|
|
|
d.
|
This Agreement, including all exhibits, constitutes the entire agreement between the parties with respect to the subject matter hereof and supersedes all proposal, negotiations and other communications between the parties, whether written or oral, with respect to the subject matter hereof.
|
|
|
e.
|
If any provisions of this Agreement shall be held to be invalid, illegal or unenforceable, the validity, legality and enforceability of the remaining provisions of this Agreement shall not be impaired thereby, and the party against whom the holding is made, shall be entitled to substitute a similar provision that preserves the benefit of the bargain.
|
|
|
f.
|
The failure of any party to insist on strict performance of any provision of this Agreement or exercise any right hereunder will not constitute a waiver of that provision or right.
|
|
|
g.
|
This Agreement may be executed in any number of counterparts, each of which shall be deemed an original as against the party whose signature appears thereon, but all of which taken together shall constitute but one and the same instrument.
|
|
|
h.
|
Each party hereto agrees to execute, acknowledge and deliver such further instruments and do all such further acts as may be necessary or appropriate in order to carry out the purposes and intent of this Agreement.
|
|
|
i.
|
The paragraph headings contained in this Agreement are for reference purposes only and shall not in any way affect the meaning or interpretation of this Agreement.
|
Rev. 16 November 2007
Page 14 of 17
IN WITNESS WHEREOF, the parties intending to be legally bound have caused this Agreement to be executed by their duly authorized representatives or, in the case of the Investigator, have duly executed this Agreement, on the dates stated beneath their names:
SITE
Accepted by:
| /s/ Xxxxxx Xxxxxxx | |
|
Name: Xxxxxx Xxxxxxx
|
|
|
Title: Vice President, Industry Relations and Research Support
|
|
| Dated: 7/24/12 |
ACTINIUM PHARMACEUTICALS, INC.
Accepted by:
| /s/ Xxxxxx Xxxxx | |
|
Name: XXXXXX XXXXX, M.D.
|
|
|
Title: CEO
|
|
| Dated: 7/26/12 |
Read and Acknowledged by:
INVESTIGATOR:
| /s/ Xxxx X. xxxxx | |
|
Name: Xxxx X. xxxxx, M.D., Ph.D.
|
|
| Dated: 7/20/12 |
Rev. 16 November 2007
Page 15 of 17
EXHIBIT A
PROTOCOL
Incorporated by reference
Rev. 16 November 2007
Page 16 of 17
EXHIBIT B
Investigator's current curriculum vitae is attached (incorporated by reference)
Rev. 16 November 2007
Page 17 of 17
EXHIBIT C
BUDGET
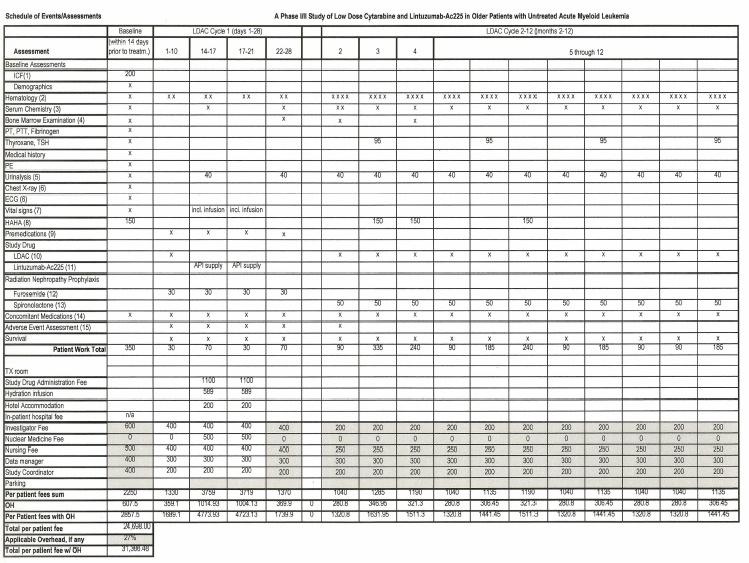

| 1 | Informed consent form (ICF) obtained prior to any study-related procedures | |
| 2 | Hematology assessments: complete blood count (CBC), Red Blood Cell count (RBC) with WBC differential and platelet count, hemoglobin, hematocrit. Timetable: Baseline, twice per week for the 1st 2 months, then CBC once every week. | |
| 3 | Serum chemistry: Comprehensive biochemistry profile (including serum electrolytes, BUN, creatinine, glucose, calcium, phosphate, total protein, albumin, alkaline phosphatase, AST, total bilirubin), PO4, LDH, and uric acid. Assessments timetable: Baseline, every other week for the 1st 2 months, then every month. | |
| 4 |
Bone marrow aspiration or biopsy: morphology, cytogenetics (if not previously done), immunophenotyping, PCR analysis if applicable. Assessment timetable: Baseline, just before cycle 2 LDAC, after the end of cycle 4 LDAC, upon disease progression or before the 2nd cycle LDAC if the peripheral blood count shows ANC > 1000 and the platelet count is > 100,000 after 225Actinium-HuM195 treatmer
|
|
| 5 | Urinalysis to include: pH, protein, ketones, Hb or blood, specific gravity and if abnormal, microscopic a examination of the sediment. Timetable: every other week for the 1st month, then monthly. | |
| 6 | ECG and Echocardiogram: within 30 days of study entry | |
| 7 |
Xxxxx xxxxx to include temperature, pulse rate, respiration rate, blood pressure. Timetable: 15 minutes prior to each study drug infusion, every 15 minutes during the infusion, then 30, 60, 90 and 120 minutes after the infusions are completed.
|
|
| 8 | Human anti-human antibody response: Baseline, then 2, 3 and 6 months after the last dose of study drug. Please include only payments for collecting and shipping from your site, as the tests will be done centrally. | |
| 9 | Allopurinol, 300-600 mg po/day one day before administration of Lintuzumab-Ac225 and continued for 7 days after the 2nd (last) dose of study drug. For subjects with ANC500/pL at study start, prophylactic antibiotic and antifungal therapy should be used. | |
| 10 | First cycle of LDAC, 20 mg subQ every 12hrs X 10 D, given prior to the administration of study drug, then cycle 2 given 3 to 4 weeks after the 2nd dose of study drug for up to 12 cycles total. | |
| 11 | 1st divided dose given 4-7 days after completion of LDAC cycle 1. 2nd divided dose given 4-7 days after dose #1 | |
| 12 | 40 mg po/day starting 1 day before LDAC administration and continuing until the end of cycle 1 (day 28) | |
| 13 | 25 mg po/day starting 1 day after the last dose of furosemide at the end of cycle 1. | |
| 14 | Concomitant medications: all medications taken for 28 days prior to the 1st dose of study drug and then all medications while on study | |
| 15 | Adverse event assessments begin at the time of LDAC administration and continue until 30 days after the last dose of the study drug. |

