SPONSORED RESEARCH AGREEMENT
Exhibit 10.26
This Agreement is entered into by Surgi-Vision Inc., incorporated in the State of Delaware (“SVI”), and the Regents of the University of California on behalf of its San Francisco campus (“UCSF”), with an administrative office located at 000 Xxxxx Xxxxxx, Xxxxx 0000, Xxx Xxxxxxxxx, Xxxxxxxxxx, 00000-0000, the above named entities hereinafter identified together as “the Parties” and in the singular as “the Party”.
In view of the foregoing, the Parties agree to the following terms of this Agreement.
1.1 In connection with work performed pursuant to this Agreement, the Parties may find it necessary or desirable to disclose to the each other certain proprietary and confidential information relating to product concepts, operation, ideas, and developments (defined below as the “Confidential Information”).
1.2 “Confidential Information” means information disclosed by one Party to another that has value to the formation and operation of the disclosing Party’s business, which is marked “Confidential”, or if orally disclosed, reduced to writing within thirty (30) days of disclosure. Notwithstanding the foregoing, “Confidential Information” shall not include information which: (a) is in the public domain when received from a Party; or (b) was known to a Party prior to its receipt from the other party, as shown by written records in existence prior to such disclosure; or (c) is independently developed by one Party as evidenced by its written records; or (d) is required to be disclosed by law. No Party shall be liable under this Agreement for disclosure or use of Confidential Information which: (i) is published or otherwise enters the public domain through no fault of the receiving party; or (ii) was lawfully obtained by the receiving party from a third party entitled to disclose it.
2
1.4 UCSF is free to publish or otherwise disclose activities performed or data arising from activities performed under this Agreement. However, UCSF and Researchers must first provide a review copy of a planned disclosure to a third party to SVI at least thirty (30) days prior thereto to allow sufficient time to review the document/planned disclosure to confirm no SVI Confidential Information is included, that SVI technology is correctly described and/or allow SVI, at its discretion, to request that patent applications be pursued for inventions that may be described in the document/planned disclosure. In no event shall the delay to publish exceed a total of sixty (60) days.
2.1 UCSF agrees that the Researchers shall reasonably carry out research activities substantially as described in the Project Plan attached at Appendix A and to cooperate with SVI to facilitate a timely and successful completion of the Project Plan. The Project Plan describes the activities to be carried out under this Agreement, including: (a) Continued Clinical Assessment of Efficacy and Safety of IMR Guidance; and (b) Advanced Technology Assessment, including an integrated head-holder and a new aiming device.
2.2 UCSF and/or its Researchers will give SVI periodic reports on the status of the Project Plan and promptly notify SVI on the date of the first clinical IMR DBS placement procedure on a human patient conducted after the Effective Date of this Agreement. The notification will be used to determine the term of the Agreement as provided below in section 6.1.
2.3 UCSF and the Researchers agree to comply with all appropriate regulations in carrying out research activities under this Agreement, including all medical and human study protocols, and FDA and other appropriate rules and regulations.
3.1 SVI agrees to provide to UCSF funding in the amount of $[***] to be allocated and applied by UCSF and the Researchers to carry out the goals and activities described in the Project Plan.
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
3
[***]. The monies will be made payable to “The Regents of the University of California” and transmitted to the address below unless such address is updated in writing by UCSF.
University of California San Francisco
UCSF Accounting - EMF
0000 Xxxxxx Xxxxxx, Xxxxx 000
Xxx Xxxxxxxxx, XX 00000-0000
3.2 SVI agrees to provide technical assistance and cooperate with Researchers to facilitate the goals and actions described in the Project Plan.
4.1 SVI and UCSF and its’ Researchers are independent contractors for all purposes of this Agreement. Neither UCSF or Researchers or any agent, representative, contractor or employee of UCSF will be considered an agent, representative or employee of SVI for any purpose. Conduct, direction and control of the work performed under this Agreement by UCSF and Researchers lies solely with same.
5.1 “Intellectual Property” means any inventions made in the direct performance of the Project Plan.
5.2 The Parties will each have the right to use data generated from the direct performance of the Project Plan.
5.3 For SVI wholly-owned Intellectual Property, SVI shall grant to UCSF and the Researchers a royalty-free, non-exclusive license to practice the technology for non commercial research purposes only.
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
4
5.4 For UCSF wholly-owned Intellectual Property, UCSF shall grant to SVI a royalty-free, non-exclusive license to practice the technology for research purposes only.
5.5 For USCF wholly or partially/jointly owned Intellectual Property, UCSF agrees to offer SVI the first opportunity to enter into a royalty-bearing commercial (exclusive or non-exclusive) license, as appropriate, at a commercially-reasonable royalty rate. SVI may exercise such opportunity by notifying UCSF of its intent to do so, within ninety (90) days of written notice by UCSF of such Intellectual Property, such notice will be no earlier than after a patent application for the invention(s) has been filed. Any exclusive license shall allow the Researchers and UCSF the ability to practice the technology covered by the license or assignment for research purposes. Should UCSF and SVI be unable to agree to terms for such commercial license or assignment within one hundred and eight days (180) days from SVI’s exercise of the opportunity, UCSF shall be free to negotiate with a third party. However, should UCSF reach a provisional agreement with any such third party within ninety days (90) of UCSF and SVI failing to come to agreement on terms for a commercial license, as described herein, SVI will have a thirty (30) day period from the receipt of notice of the third party provisional agreement, to exercise a right of first refusal on financial terms and conditions as set forth in such proffered third party agreement.
5.6 In the event of a joint invention resulting in co-owned Intellectual Property arising from this Agreement, SVI and UCSF will cooperate and mutually agree upon outside patent counsel and the preparation, filing, prosecution and maintenance of any patent applications and resulting patents covering same, including the right for both Parties to review and approve any such patent application filing.
6.1 This Agreement is effective on the last signature date of the undersigned Parties (“the Effective Date”) and continues in effect for one year from the date of a first clinical IMR DBS placement procedure on a human patient conducted after the Effective Date of this Agreement if the payments to UCSF under section 3.1 above has been made.
5
6.2 The Agreement may be extended for an additional one (1) year period by mutual written agreement of the Parties.
7.1 SVI, UCSF and Researchers contemplate that performance of activities arising from this Agreement may include joint or collaborative research and activities between the Parties and/or affiliates or successors in interest thereof. Hence, this Agreement may be asserted as a joint research agreement for the performance of experimental, developmental or research work in the field of Interventional Magnetic Resonance (IMR) deemed to have been owned by the same person or subject to an obligation of assignment to the same person under 35 USC § 103(c) as provided for in the Cooperative Research and Technology Enhancement Act of 2004.
8.1 UCSF certifies to the best of its current knowledge that there is no prior, preexisting or existing agreement with a third party that conflicts with this Agreement.
8.2 UCSF certify that it/they has/have the full right and authority to enter into this Agreement.
8.3 UCSF certifies that all of their employees (including other principal investigators, students and/or faculty), whose services may be used to carry out research and/or development activities under this Agreement, are or will be appropriately informed of the terms of this Agreement, and that all such persons are under legal obligation to UCSF by contract or otherwise, sufficient to fully comply with this Agreement, including for persons that may be inventors, legal obligations to assign rights to any inventions and associated operational copyrights to UCSF.
6
9.1 This agreement will be governed by and construed in accordance with the laws of the State of California. Every provision of this agreement is intended to be severable. If any term or provision hereof is illegal or invalid for any reason whatsoever, such illegality or invalidity shall not affect the remainder of this agreement.
| To UCSF | To Surgi-Vision, Inc.: | |
| Attn: Director | Xxxxxx Xxxxxxx | |
| University of California at San Francisco | President & CEO | |
| 000 Xxxxx Xxxxxx, Xxxxx 0000 | 00 Xxxxx Xxxxx St.; 00xx Xxxxx | |
| Xxx Xxxxxxxxx, XX 00000-0000 | Xxxxxxx, XX 00000 | |
7
| On behalf of USCF | On behalf of Surgi-Vision, Inc. | |||
| /s/ Xxx Xxxxxxxx | /s/ Xxxxxx Xxxxxxx | |||
| Xxx Xxxxxxxx | Xxxxxx Xxxxxxx, President and CEO | |||
| Industry Contracts Manager Office of Sponsored Research University of California San Francisco |
||||
| Date: 8/15/07 | Date: 8/24/07 | |||
| Read and Understood: | ||||
| /s/ Xxxxxxx Xxxxx | 8/13/07 | |||
| Xxxxxxx Xxxxx, M.D. | (Date) | |||
| /s/ Xxxx Xxxxxx | 8/10/07 | |||
| Xxxx Xxxxxx, M.D. | (Date) | |||
| /s/ Xxxxxxxx Xxxxxx | 8/10/07 | |||
| Xxxxxxxx Xxxxxx, Ph.D. | (Date) | |||
8
Appendix A
Project Plan
9
Surgi-Vision Scope of Work & Budget Justification
“MRI Implanation for Deep Brain Stimulation (DBS)”
UCSF Investigators
Drs. Xxxxxx Xxxxx, Xxxx Xxxxxx, Xxxxxxxx Xxxxxx & Xxxx Xxxxxx
Xxxxx 0, 0000-Xxxxxxxx 00, 0000
XXXXX OF WORK
Phase 1 - Assessment
Clinical Assessment of Efficacy and Safety of IMR Guidance
Perform IMR guided DBS lead insertions in Xxxxxxxxx’x and Dystonia patients. Estimate approximately 15 patients in the first year, with 3 hrs/case research MR time required.
Advanced Technology Assessment
This section will involve two principle components:[***]. Evaluations will initially be performed in phantoms and compared with prior accuracy and reproducibility testing. CHR approval has been established for use in human pending these initial findings.
Phase 2 - Initiation of New Development Projects
The work in this phase will involve development of new surgical tools that will be tested in the MR scanners with plastic, phantom heads. The initial work will basically be “bench research” with experiments done in the scanner to check accuracy and feasibility of use.
Preliminary Design of an Integrated Head-Holder
Implementation of iMRI across multiple MR platforms will require a head-holder that will attach to different MR gantries. [***] This prototype would be designed, manufactured, tested and implemented in clinical use within year 1.
Preliminary Design for a New Aiming Device
Current aiming technology has several shortcomings including difficulty with simultaneous bilateral implantation, relative narrow “aiming angles” with respect to the xxxx hole and awkward manipulation of the devices with the patient at isocenter. Solutions to these issues and preliminary solutions and possible designs for new aiming technologies will be explored in year 1. Review of current agreements with existing entities is required to avoid potential conflict,
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
10
FIRST AMENDMENT TO
This First Amendment to Sponsored Research Agreement (the “Amendment”) is made effective as of December 1, 2008 (the “Amendment Effective Date”), by and between SurgiVision, Inc. (f/k/a Surgi-Vision, Inc.), a Delaware corporation (“SVI”), and the Regents of the University of California on behalf of its San Francisco campus (“UCSF”).
WHEREAS, SVI and UCSF entered into that certain Sponsored Research Agreement in August 2007 (the “Research Agreement”); and
1. Defined Terms. Capitalized terms used but not defined in this Amendment shall have the meanings ascribed to such terms in the Research Agreement.
2. Term of Research Agreement. Subject to Section 4 below, the term of the Research Agreement shall continue through April 30, 2009 (the “Expiration Date”).
3. Additional SVI Support. In addition to the funding described in Section 3.1 of the Research Agreement (which was paid by SVI to UCSF in accordance with the Research Agreement), SVI agrees, subject to Section 4 below, to provide to UCSF funding in an amount up to $[***]. Such funding shall be allocated and applied by UCSF (a) to carry out research activities under the Research Agreement during the 5-month period commencing with the Amendment Effective Date and continuing through the Expiration Date, and (b) substantially in accordance with the itemized budget attached hereto as Appendix A. SVI shall remit monthly payments to UCSF based on monthly invoices submitted to SVI by UCSF. Such invoices shall itemize the direct costs and identify the facility and administrative costs. Invoices submitted to SVI shall be paid by SVI within 30 days of receipt.
4. UC Discovery Grant. UCSF acknowledges that (a) SVI is the industry sponsor for a research proposal entitled “Optimized Methodology for Implantation of DBS Electrodes” (Principal Investigator: Xxxxxxxx X. Xxxxxx, Ph.D.) submitted pursuant to the UC Discovery Grant Request for Proposals, and (b) if that proposed project is approved for UC Discovery Grant funding by the Industry-University Cooperative Research Program, SVI intends to negotiate with UCSF with the goal of executing a mutually acceptable research agreement (the “UC Discovery Agreement”). If SVI and UCSF enter into the UC Discovery Agreement, then the term of the Research Agreement shall expire as of the effective date of the UC Discovery Agreement and SVI shall not be obligated to provide funding under the Research Agreement for any period of time beyond that date. Notwithstanding the expiration of the Research Agreement, the Intellectual Property, Joint Research and Confidentiality provisions of the Research Agreement shall continue to apply as otherwise provided in Section 6.3 of the Research Agreement.
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
1
5. Notice to SVI. SVI’s address for notice under the Research Agreement is:
SurgiVision, Inc.
Attention: CEO
Xxx Xxxxxxxx Xxxxxx
Xxxxx 0000
Xxxxxxx, XX 00000
with a copy to:
SurgiVision, Inc.
Attention: VP, Business Affairs
Xxx Xxxxxxxx Xxxxxx
Xxxxx 0000
Xxxxxxx, XX 00000
[The next page is the signature page]
2
| On behalf of UCSF | On behalf of SVI | |||
| /s/ Kent Iwamlya | /s/ Xxxxx Xxxxxx | |||
| Xxxx Iwamlya | XXXXX XXXXXX | |||
| Industry Contracts Officer | VICE PRESIDENT, BUSINESS AFFAIRS | |||
| Office of Sponsored Research University of California San Xxxxxxxxx |
||||
| Date: 2/25/09 | Date: February 16, 2009 | |||
| Read and Understood: | ||||
| /s/ Xxxxxxx Xxxxx | 2/17/09 | |||
| Xxxxxxx Xxxxx, M.D. | Date | |||
| /s/ Xxxx Xxxxxx | 2/18/09 | |||
| Xxxx Xxxxxx, M.D. | Date | |||
| /s/ Xxxxxxxx Xxxxxx | 2/24/09 | |||
| Xxxxxxxx Xxxxxx, Ph.D. | Date | |||
3
Appendix A
Budget
[See Attached]
4
[***]
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
5
SECOND AMENDMENT TO
This Second Amendment to the Sponsored Research Agreement (“Second Amendment”) is made effective as of May 1, 2009 (the “Second Amendment Effective Date”) by and between SurgiVision, Inc. (f/k/a Surgi-Vision, Inc.), a Delaware corporation (“SVI”), and The Regents of the University of California on behalf of its San Francisco campus (“UCSF”).
A. SVI and UCSF entered into a Sponsored Research Agreement in August 2007, which was subsequently amended effective as of December 1, 2008 (as amended, the “Research Agreement”).
B. SVI and UCSF wish to further amend the terms of the Research Agreement as set forth below.
NOW, THEREFORE, for good and valuable consideration, the sufficiency and receipt of which are hereby acknowledged, it is hereby agreed as follows:
1. Capitalized terms used but not defined in this Second Amendment shall have the meanings ascribed to such terms in the Research Agreement.
2. Subject to Section 5 below, the term of the Research Agreement shall continue through April 30, 2010 (the “Expiration Date”).
3. Subject to Section 5 below, for the one-year period commencing with the Second Amendment Effective Date and continuing through the Expiration Date, SVI shall provide to UCSF funding in an amount up to $[***] (the “Additional Funding”). The Additional Funding shall be allocated and applied by UCSF (a) to carry out research activities substantially as described in the Scope of Work attached hereto as Exhibit A (the “SOW”), and (b) substantially in accordance with the itemized budget attached hereto as Exhibit B. Subject to Section 5 below,
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
1
SVI shall pay to UCSF the Additional Funding in [***] each according to the following schedule: [***]. For purposes of the Research Agreement (as amended by this Second Amendment), the term “Project Plan” shall hereinafter include, without limitation, the SOW attached hereto as Exhibit A.
4. Installment payments of the Additional Funding shall be made payable to “The Regents of the University of California” and transmitted to the address below unless such address is updated by written notice to SVI from UCSF:
University of California San Francisco
UCSF Accounting – EMF
0000 Xxxxxx Xxxxxx, Xxxxx 000
Xxx Xxxxxxxxx, XX 00000-0000
5. UCSF acknowledges that (a) SVI is the industry sponsor for a research proposal entitled “Optimized Methodology for Implantation of DBS Electrodes” (Principal Investigator: Xxxxxxxx X. Xxxxxx, Ph.D.) submitted pursuant to the UC Discovery Grant Request for Proposals, and (b) if that proposed project is approved for UC Discovery Grant funding by the Industry-University Cooperative Research Program, SVI intends to negotiate with UCSF with the goal of executing a mutually acceptable research agreement (the “UC Discovery Agreement”). If SVI and UCSF enter into the UC Discovery Agreement, then (x) the term of the Research Agreement shall expire as of the effective date of the UC Discovery Agreement, (y) SVI shall not be obligated to pay any further installments of the Additional Funding, and (z) UCSF shall promptly return to SVI that portion of any installment of the Additional Funding paid by SVI that is attributable to the period of time that follows the expiration of the Research Agreement. Notwithstanding the expiration of the Research Agreement, the Intellectual Property, Joint Research and Confidentiality provisions of the Research Agreement shall continue to apply as otherwise provided in Section 6.3 of the Research Agreement.
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
2
5. Section 5 of the Research Agreement (Intellectual Property) is hereby amended by adding the following section 5.7:
“5.7 Notwithstanding the provisions of section 5.5 above to the contrary, with respect to any UCSF wholly or partially/jointly owned Intellectual Property that is dominated by patent rights (whether pursuant to an issued patent or pending patent application) currently owned or controlled by SVI (“Dominated IP”), UCSF hereby grants to SVI an irrevocable fully paid-up, non-royalty bearing, worldwide non-exclusive license, with the right to sublicense, under the Dominated IP to make, have made, use, import, offer for sale and sell products and processes covered by the Dominated IP. UCSF shall, upon SVI’s written request, file patent application(s) for any such Dominated IP, provided that SVI shall reimburse UCSF for the prosecution costs and expenses incurred by UCSF with respect to any such application(s) requested by SVI.”
6. UCSF shall provide SVI with information reasonably requested by SVI relating to any clinical procedures performed using SVI’s DBS implantation platform as contemplated in the SOW, except information that is subject to patient confidentiality laws or that UCSF is otherwise prohibited from providing to SVI pursuant to applicable law.
7. The Exhibits attached to this Second Amendment are hereby incorporated into and made a part of this Second Amendment.
8. Except as expressly provided in this Second Amendment, all other terms, conditions and provisions of the Research Agreement shall continue in full force and effect as provided therein.
[The next page is the signature page]
3
IN WITNESS WHEREOF, SVI and UCSF have entered into this Second Amendment to be effective as of the date first set forth above.
| THE REGENTS OF THE UNIVERSITY OF CALIFORNIA |
SURGIVISION, INC. | |||||||
| By | /s/ Kent Iwamlya | By | /s/ Xxx Xxxxxxx | |||||
| Name: | Kent Iwamlya | Name: | XXX XXXXXXX | |||||
| Title: | Industry Contracts Officer Office of Sponsored Research University of California San Francisco |
Title: | CEO | |||||
| Date: | 7-10-09 | Date: | 7/15/2009 | |||||
Each of the undersigned Researchers, while not a party to this Second Amendment, hereby acknowledges that he has read the Second Amendment and understands his obligations as an UCSF employee hereunder:
| /s/ Xxxxxxxx Xxxxxx | ||||||||
| Name: | Xxxxxxxx Xxxxxx, PhD | |||||||
| Date: | July 7, 2009 | |||||||
| /s/ Xxxxxxx Xxxxx | ||||||||
| Name: | Xxxxxxx Xxxxx | |||||||
| Date: | July 9, 2009 | |||||||
| By | ||||||||
| Name: | ||||||||
| Date: | ||||||||
4
Exhibit A
Scope of Work
[See Attached]
5
Surgi-Vision Scope of Work & Budget
Justification
“MRI Implanation for Deep Brain Stimulation (DBS)”
UCSF Investigators
Drs. Xxxxxx Xxxxx, Xxxx Xxxxxx, Xxxxxxxx Xxxxxx & Xxxx Xxxxxx
May 1, 2009 - April 30, 2010
SCOPE OF WORK
Phase 1 – Clinical Assessment of Safety and Efficacy of IMR Guidance
Perform IMR guided DBS lead insertions in Xxxxxxxxx’x and Dystonia patients. We anticipate performing 12 patients during the term of this agreement, with 3 hrs/case research MR time required. Implantations during Phase 1 will continue to utilize the original prototype system until acceptable testing of the Surgi-Vision system (Phase 2) has been achieved. Patients will undergo comprehensive neurological evaluations prior to surgery and following stimulation and optimization.
Phase 2 – Phantom Evaluation of Surgi-Vision Delivery System
Surgi-Vision has developed a complete DBS implantation platform including an RF coil, head fixation frame, trajectory guide, and comprehensive implantation software. This delivery system is presently undergoing FDA review and may be certified by Fall, 2009. [***]
Phase 3 – Transition to clinical utilization of Surgi-Vision Delivery System
In the Fall of 2009 we will begin performing DBS implantations in PD and Dystonia patients with the new Surgi-Vision platform. This will either be under IRB approval based on our phantom evaluations (Phase 2) or with FDA approved product, depending on the pace or regulatory approval. [***]
Phase 4 – Advanced Technology Assessment
This section will involve two principle components: [***] Evaluations will initially be performed in phantoms and compared with prior accuracy and reproducibility testing. CHR approval has been established for use in human pending these initial findings.
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
6
Exhibit B
Research Budget
[See Attached]
7
[***]
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
8
THIRD AMENDMENT TO
This Third Amendment to Sponsored Research Agreement (this “Third Amendment”) is made effective as of November 2, 2009 (the “Third Amendment Effective Date”) by and between SurgiVision, Inc., a Delaware corporation (“SVI”), and The Regents of the University of California on behalf of its San Francisco campus (“UCSF”).
A SVI and UCSF entered into a Sponsored Research Agreement in August 2007, as amended pursuant that certain First Amendment to Sponsored Research Agreement made effective as of December 1, 2008 and that certain Second Amendment to Sponsored Research Agreement made effective as of May 1, 2009 (as amended, the “Research Agreement”).
B. UCSF submitted a research proposal entitled “Optimized Methodology for Implantation of DBS Electrodes” (Principal Investigator: Xxxxxxxx X. Xxxxxx, Ph.D.) pursuant to the UC Discovery Grant Request for Proposals (the “Research Project”).
C. The Research Project has been approved for UC Discovery Grant funding by the Industry-University Cooperative Research Program.
D. The Second Amendment to Sponsored Research Agreement made effective as of May 1, 2009 (the “Second Amendment”) contemplated that upon approval of the Research Project for UC Discovery Grant funding, SVI and UCSF would negotiate a new UC Discovery Agreement, which agreement would replace the Research Agreement.
E. Notwithstanding the provisions of the Second Amendment to the contrary, in lieu of entering into the UC Discovery Agreement, SVI and UCSF wish to further amend the terms of the Research Agreement as set forth below to address the Research Project and SVI’s support with respect thereto.
NOW, THEREFORE, for good and valuable consideration, the sufficiency and receipt of which are hereby acknowledged, it is hereby agreed as follows:
1. Defined Terms. Capitalized terms used but not defined in this Third Amendment shall have the meanings ascribed to such terms in the Research Agreement.
2. Term of Research Agreement.
(a) Unless terminated earlier as provided below, the term of the Research Agreement shall continue through November 1, 2011 (the “Expiration Date”).
1
(b) If either SVI or UCSF (or any Principal Investigator) materially defaults in the performance of any duty or obligation imposed upon it under the Research Agreement (as amended by this Third Amendment) and such default continues for sixty (60) days after written notice thereof has been given to the defaulting party by the other party, such other party may (but need not) give notice of the immediate termination of the Research Agreement.
3. SVI Support for Research Project.
(a) SVI’s funding obligations under Section 3 of the Second Amendment are hereby terminated.
(b) With respect to the two-year period commencing with the Third Amendment Effective Date and continuing through the Expiration Date, SVI shall provide to UCSF funding in an aggregate amount up to $[***] (the “Cash Funding”). UCSF shall allocate and apply the Cash Funding (i) to carry out research activities for the Research Project substantially as described in the Scope of Work attached hereto as Exhibit A (the “Research Project SOW”), and (ii) substantially in accordance with the itemized budget for the Research Project attached hereto as Exhibit B. SVI shall pay to UCSF the Cash Funding in [***] according to the following schedule: [***]. For purposes of the Research Agreement (as amended by this Third Amendment), the term “Project Plan” shall hereinafter include, without limitation, the Research Project SOW attached hereto as Exhibit A.
(c) In addition to the Cash Funding, and as further support for the Research Project, SVI shall make the in-kind contributions to UCSF set forth on Exhibit C attached hereto (the “In-Kind Contributions”). UCSF and SVI acknowledge and agree that the value of the In-Kind Contributions, as reflected on Exhibit C, will be amortized over the two-year period commencing with the Third Amendment Effective Date and continuing through the Expiration Date, resulting in an annual valuation of the In-Kind Contributions equal to $[***]. UCSF shall use the In-Kind Contributions (i) to carry out research activities for the Research Project substantially as described in the Research Project SOW, and (ii) with appropriate care in accordance with all instructions for use and in compliance with applicable law.
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
2
(d) Installment payments of the Cash Funding shall be made payable to “The Regents of the University of California” and transmitted to the address below unless such address is updated by written notice to SVI from UCSF:
University of California San Francisco
UCSF Accounting – EMF
0000 Xxxxxx Xxxxxx, Xxxxx 000
Xxx Xxxxxxxxx, XX 00000-0000
(e) In the event the Research Agreement is terminated by SVI pursuant to Section 2(b) above prior to the Expiration Date, UCSF shall promptly return to SVI that portion of any installment of the Cash Funding paid by SVI that is attributable to the period of time that follows the termination of the Research Agreement.
4. Human Subject Research. UCSF and the Principal Investigators understand that the research covered by the Research Agreement involving human subjects (“Human Subject Research”) requires appropriate documentation, review and approval by UCSF’s Institutional Review Board (the “IRB”) and compliance with all IRB recommendations and requirements. UCSF and the Principal Investigators acknowledge and agree that (a) a copy of each IRB approval relating to Human Subject Research covered by the Research Agreement will be provided to SVI, (b) all clinical studies will be conducted under the supervision of qualified and licensed physicians, (c) all FDA regulations for Human Subject Research will be strictly observed, and (d) no Human Subject Research will be commenced before IRB approval has been granted. UCSF and the Principal Investigators assume full responsibility for any clinical decisions made as a result of data, directly or indirectly, generated during any research covered by the Research Agreement.
5. Clinical Data.
(a) UCSF shall provide SVI with information reasonably requested by SVI relating to any clinical procedures performed using SVI’s ClearPointTM Neuro Intervention System as contemplated in the Research Project SOW, except information that is subject to patient confidentiality laws or that UCSF is otherwise prohibited from providing to SVI pursuant to applicable law.
3
(b) UCSF agrees to purge all patient identifiers from all information it provides to SVI hereunder. Nevertheless, and to the extent required by the provisions of the Health Insurance Portability and Accountability Act (“HIPAA”) and the regulations promulgated thereunder, SVI does hereby assure UCSF that it will appropriately safeguard protected health information (“PHI”) made available to or obtained by SVI hereunder. Without limiting the obligations of SVI otherwise set forth herein or imposed by applicable law, SVI agrees to comply with applicable requirements of law relating to PHI. Specifically, SVI shall:
(i) not use or disclose PHI other than as permitted or required by the Research Agreement (including this Third Amendment) or as permitted or required by law;
(ii) implement administrative, physical and technical safeguards that reasonably and appropriately protect the confidentiality, integrity and availability of the electronic PHI that it creates, receives, maintains or transmits on behalf of UCSF and use appropriate safeguards to prevent use or disclosure of PHI other than as provided for herein;
(iii) report to UCSF any use or disclosure of PHI not provided for herein, and any security incident relating to PHI, of which SVI becomes aware;
(iv) ensure that any subcontractors or agents to whom SVI provides PHI received from, or created or received by SVI on behalf of, UCSF agree to essentially the same restrictions and conditions that apply to SVI with respect to PHI and implement reasonable and appropriate safeguards with respect to such information;
(v) make PHI available to UCSF in accordance with applicable law;
(vi) permit UCSF to access PHI to make or permit others to make amendments to PHI in accordance with applicable law;
(vii) make available to UCSF the information in SVI’s possession required to provide an accounting of SVI’s disclosures of PHI as required by applicable law;
(viii) make SVI’s internal practices, books, and records relating to the use and disclosure of PHI received from UCSF available to the Secretary of the United States Department of Health & Human Services for purposes of determining UCSF’s compliance with applicable law;
(ix) use reasonable commercial efforts to mitigate any harmful effect that is known to SVI of a use or disclosure of PHI by SVI in violation of the requirements set forth herein; and
(x) upon expiration or termination of the Research Agreement, return to UCSF or destroy all PHI in its possession as a result of this Amendment and retain no copies of such PHI, if it is feasible to do so. If return or destruction is not feasible, SVI agrees to extend all protections contained here to SVI’s use and/or disclosure of any retained PHI, and to limit further uses and/or disclosures to the purposes that make the return or destruction of the PHI infeasible.
4
(c) SVI agrees that it will negotiate in good faith an amendment hereto if required by, and to the extent required by, the provisions of HIPAA and regulations promulgated thereunder, in order to assure that this Amendment is consistent therewith.
6. Prohibition on Practice of Medicine . Notwithstanding anything to the contrary contained herein, the parties acknowledge that SVI is not authorized or qualified to engage in any activity which may be construed or deemed to constitute the practice of medicine. Accordingly, UCSF shall retain the authority to direct all medical decisions regarding the care and treatment of its patients and shall assume full responsibility for any clinical decisions made as a result of data, directly or indirectly, generated during the research activities conducted. SVI shall neither exercise control over nor interfere with the physician-patient relationship. To the extent any act or service required of SVI under the Research Agreement should be construed or deemed by a governmental authority, agency or court to constitute the practice of medicine, the performance of said act or service by SVI shall be deemed waived and forever unenforceable.
7. Anti-Kickback Statute. In compliance with the federal Medicare/Medicaid Anti-Kickback Statute, each party represents that the Cash Funding and In-Kind Contributions to UCSF have not been determined with regard to any implicit or explicit agreement to provide favorable procurement decisions with regard to SVI’s products, and have not been given in exchange for such decisions. Each party further represents that such compensation has not been determined with regard to the value or volume of any business generated between the parties and that such compensation is consistent with fair market value in arm’s length transactions. The compensation provided hereunder is directly related to the costs of carrying out research, and includes no incentive payment to any individual for identifying or recruiting human subjects. The Research Agreement (including this Third Amendment) is not intended to, and does not, induce the referral of patients or to induce purchase of any items or services reimbursed by any federal or state health care program. UCSF acknowledges that (a) it may be obligated to report the “no-charge” status of the In-Kind Contributions to Medicare, Medicaid and/or other federal health care programs, and (b) it may also have reporting obligations to third parties (including, without limitation, Medicare) that require the allocation or classification of the In-Kind Contributions in accordance with particular reporting principles. UCSF agrees that it is solely responsible for any such reporting, allocation(s) and/or classification(s).
8. FDA Regulations. UCSF understands and acknowledges that, as of the Third Amendment Effective Date, pending 510(k) marketing clearance from the U.S. Food and Drug Administration (“FDA”), the In-Kind Contributions are not available for sale in the United States. Accordingly, notwithstanding any provision herein to the contrary, pending 510(k) marketing clearance from the FDA, UCSF and the Principal Investigators shall use the In-Kind Contributions only to the extent such use is permitted under FDA regulations. Furthermore, pending 510(k) marketing clearance from the FDA, UCSF agrees that it will negotiate in good faith an amendment hereto if required by, and to the extent required by, FDA regulations in order to assure that this Amendment is consistent therewith.
5
9. Exhibits. The Exhibits attached to this Third Amendment are hereby incorporated into and made a part of this Third Amendment.
10. Ratification of Research Agreement. Except as provided in this Third Amendment, all other terms, conditions and provisions of the Research Agreement shall continue in full force and effect as provided therein.
[The next page is the signature page]
6
IN WITNESS WHEREOF, SVI and UCSF have entered into this Third Amendment to be effective as of the date first set forth above.
| THE REGENTS OF THE UNIVERSITY OF CALIFORNIA |
SURGIVISION, INC. | |||||||
| By | /s/ Xxx Xxxxxxxx | By | /s/ Xxx Xxxxxxx | |||||
| Name: | Xxx Xxxxxxxx | Name: | Xxx Xxxxxxx | |||||
| Industry Contracts Manager Office of Sponsored Research University of California | Title: | CEO | ||||||
| Title: | San Francisco | |||||||
| Date: | Oct. 30, 2009 | Date: | Oct. 30, 2009 | |||||
Each of the undersigned Researchers, while not a party to this Third Amendment, hereby acknowledges that he has read the Third Amendment and understands his obligations as an UCSF employee hereunder:
| /s/ Xxxxxxxx X. Xxxxxx | ||||||||
| Name: Xxxxxxxx X. Xxxxxx, PhD | ||||||||
| Date: November 2, 2009 | ||||||||
| Name: | ||||||||
| Date: | ||||||||
| By | ||||||||
| Name: | ||||||||
| Date: | ||||||||
7
Exhibit A
Scope of Work
[See Attached]
8
Project Participants, Roles and % Effort
| Name, Title, Institution |
Degrees | Role on Project | % Effort | Incl.
in Budget (Yes/No) |
[***]
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
9
RESEARCH PLAN:
Discovery Research and Training (DRT) Grant
Deep brain stimulation (DBS) is a therapy that is presently primarily applied to movement disorders, but has shown promise in a number of other neurological conditions. In order for DBS therapy to provide maximum benefit with minimal side effects, electrodes must be precisely positioned at specific structures in the brain. The present methodology for implantation of DBS electrodes is to employ stereotactic methods, which are seeded with pre-operative magnetic resonance (MR) or computed tomography (CT) images. Unfortunately, stereotaxy alone does not provide the accuracy necessary for DBS therapy and thus intra-operative physiologic tests are commonly employed to indirectly infer position prior to electrode insertion. These tests are invasive, time consuming and require the patient to be awake and off their movement disorder medications during surgery.
We are pioneering a novel technique for implanting DBS electrodes with intra-operative MR guidance. The use of intra-operative MR imaging has several significant advantages, including the ability to directly visualize the deep brain target and confirm technical success prior to completion of surgery. It may obviate the need for invasive physiologic tests on awake patients and therefore open the therapy up to a wider range of patients and clinical conditions. It further has the potential to substantially simplify and shorten the operative procedure, minimize the risk of hemorrhagic complications, and result in more accurate and consistent DBS electrode positioning. Our approach, however, essentially precludes the acquisition of intra-operative physiologic evaluations of the patient. We must therefore be able to demonstrate that clinical efficacy is not compromised if anatomically favorable positioning is achieved without supporting physiologic evidence.
In this proposal we aim to develop and validate a delivery system that is optimized for implantation of DBS electrodes with MR guidance. We will partner with Surgi-Vision, Inc, a small California-based company whose mandate is to harness the power of MR imaging to drive the next generation of minimally invasive surgeries. We propose to develop a delivery system and validate its performance initially in phantom models and subsequently in patients receiving DBS therapy for movement disorders. Efficacy will be evaluated based on both technical (implantation accuracy) and clinical (benefits of stimulation) measures.
The specific aims of this proposal are as follows:
| (1) | Develop and validate an optimized trajectory guide, MR receiver coil, software interface, and imaging methodologies for localizing deep brain structures with MR imaging. |
Hypothesis: The developed delivery system will be capable of localizing targets at depths comparable to deep brain structures with sub-mm accuracy. This will be evaluated in an anatomically realistic phantom.
| (2) | Determine the implantation accuracy that can be achieved in patients receiving DBS electrodes in the subthalamic nucleus for treatment of Xxxxxxxxx’x disease. |
Hypothesis: Highly accurate positioning of DBS electrodes will be possible in vivo with intra-operative MR guidance; targeting accuracies comparable to those obtained in phantoms will be realized.
| (3) | Determine the implantation accuracy that can be achieved in patients receiving DBS electrodes in the globus pallidus interna for treatment of Dystonia. |
Hypothesis: The targeting accuracy that was achieved in the STN of DBS patients can be reproduced when targeting alternate deep brain structures.
| (4) | Assess the clinical efficacy of image guided DBS implantation, without supporting intra-operative physiologic evaluations, for treatment of Xxxxxxxxx’x disease. |
Hypothesis: implantation of DBS electrodes with intra-operative image guidance, but without intra-operative physiologic feedback, will provide clinical outcomes equivalent or superior to that obtained with conventional implantation methods.
Successful achievement of these specific aims will create a new paradigm for accessing deep brain structures with high precision and should allow shorter and safer operative procedures. Moreover, the techniques developed will be directly applicable to future therapies, such as localized drug or gene therapy and cell transplantation, where precise and minimally invasive delivery will be crucial to therapeutic efficacy. It will promote the goals of the UC Discovery Program by supporting an alliance between UC faculty and the private sector to develop an efficient, minimally invasive means of delivering a rapidly expanding therapy.
10
2. Background, Significance and Preliminary Studies
Movement disorder therapy has progressed significantly over the past decade. Pharmacologic therapies are established as the initial treatment for the vast majority of patients suffering from movement disorders. However, in severe cases, or where the disease progresses to a point where symptoms can no longer be adequately managed with medications, surgical intervention is increasingly being applied. Surgical interventions have undergone a transition from permanent lesioning of brain structures to the use of chronic stimulation electrodes whose effects are adjustable and reversible. Several structures within the thalamus and basal ganglia have been targeted, including the ventrolateral thalamus, dorsolateral sub-thalamic nucleus (STN), and posterior globus pallidus interna (GPI). The technology has primarily been applied to the treatment of Xxxxxxxxx’x disease (1) although other movement disorders including essential tremor (2) and dystonia (3) also appear to benefit from stimulation therapy. Additionally, DBS therapy has shown promise for an expanding list of applications including epilepsy (4), Tourette’s syndrome (5), obsessive compulsive disorders (6), depression (7), and traumatic brain injury (8).
Magnetic resonance (MR) imaging plays a key role in the application and evaluation of these therapies. Specific deep brain structures can either be directly visualized with MR techniques, or can be inferred based on the location of surrounding structures. These capabilities make MR the modality of choice for pre-operative planning and post-operative assessment of electrode positioning.
In this application, we propose to expand on our preliminary work aimed at using direct MR image guidance to place DBS electrodes within specific targets in the basal ganglia. Through our partnership with Surgi-Vision, Inc, we plan to create an optimized platform for precisely targeting deep brain structures. We will validate the methodology by assessing targeting accuracy in both phantoms and in two separate deep brain structures (STN and GPI). In order to assure that we are able to demonstrate clinical efficacy that is comparable to conventional implantation methodologies, we will evaluate clinical outcomes by measuring neurological and neuropsychological factors prior to and following DBS implantation. The technique has numerous potential advantages including more consistent electrode positioning, shorter surgeries, fewer brain penetrations and the ability to anesthetize patients during surgery.
2.1 MOVEMENT DISORDERS AND DEEP BRAIN STIMULATION
Movement disorders are neurological conditions that affect approximately 6 million people in the United States. Movement disorders of basal ganglia origin are characterized by either excessive movement (hyperkinetic disorders) or a lack of movement (hypokinetic disorders) and can be extremely debilitating. The appropriateness of treating a specific target within the basal ganglia is highly dependant on both the type of movement disorder and the nature of the therapy to be deployed.
2.1.1 Xxxxxxxxx’x Disease
Xxxxxxxxx’x disease (PD) is second only to Alzheimer’s as the most common neurodegenerative disease of aging. The National institutes of Health estimate that Xxxxxxxxx’x disease affects between 1,000,000 and 1,500,000 individuals in the United States, with some 20,000 new cases diagnosed each year. PD is a progressive disease and patients experience increasingly severe motor impairment including muscular rigidity, tremor, bradykinesia, difficulty with balance and other non-motor functions (9). Levodopa, in combination with carbidopa, is presently the most effective medical therapy for PD. Unfortunately, current pharmacologic therapies are associated with significant complications with long-term use and surgical interventions are considered when they begin to fail.
The severity of parkinsonian symptoms and the effectiveness of therapies can be measured using a neurological rating scale (10). The Unified Xxxxxxxxx’x Disease Rating Scale (UPDRS) is a standardized, validated rating scale that is widely used for the assessment of the severity of PD and response to therapeutic interventions (11). It consists of four subscales that evaluate mentation, behavior and mood (UPDRS-I), activities of daily living (UPDRS-II)), motor symptoms (UPDRS-III), and complications of therapy (UPDRS-IV). These categories are evaluated in an interview and examination with a neurologist and a score is established to provide a quantitative index of disease severity. A UPDRS rating of 0 corresponds to normal function while higher ratings correlate with increasingly severe or disabling symptoms. UPDRS-III specifically involves a motor examination that is typically performed in the off-medication state to determine a patient’s true parkinsonian motor symptoms, without being masked by the effects of medication. UPDRS-III is also typically
11
performed in the patient’s best on-medication state to identify what PD symptoms respond to anti-Parkinsonian therapies and the degree of the response. UPDRS scores are used widely in clinical practice to monitor disease progression and in research to assess the effects of various medications and surgical interventions for PD. Motor symptom diaries are also important to determine the degree of motor fluctuation (degree of wearing off, on-off phenomina, and dyskinesia) the patient is experiencing
2.1.2 Dystonia
Dystonia is a syndrome of sustained muscle contractions producing writhing movements and abnormal postures. It may be a primary disorder that occurs without other neurological conditions, or it may occur secondary to a central nervous system lesion whose origin may be stroke, trauma, cerebral palsy, or degenerative disease. Dystonia affects approximately 250,000 people in the U.S., making it the third most common movement disorder, following PD and Essential Tremor. Unlike PD, dystonia commonly affects children, with early onset primary dystonia typically presenting in childhood or early adolescence. While the cause of dystonia is not well understood, it is known to be a predominantly hereditary disease and is assumed to originate in motor centers within the basal ganglia. A mutation to a gene, called DYT1, has been linked to a high number of early onset dystonias.
Most forms of dystonia respond poorly to currently available systemic medications. Focal dystonias may benefit from botulinum toxin-induced denervation, but this therapy is not applicable to more generalized cases. An index of disease severity has also been established for dystonia and is referred to as the Xxxxx-Xxxx-Xxxxxxx Dystonia Rating Scale or BFMDRS (12). This Index assesses dystonia severity and frequency in nine body regions on a scale of 0-120, where a rating of 0 is again indicative of normal function. The scale is widely used for the evaluation of both adult and pediatric patients, although it has only been specifically validated for adult patients (12,13).
2.1.3 Deep Brain Stimulation in Movement Disorders
Surgical intervention is indicated when symptoms of movement disorders can no longer be adequately managed with medications. Surgical methods for suppressing movement disorders target specific nuclei within the basal ganglia and require precise access to relatively small deep brain structures where open surgical access is largely impractical. The surgical aim is to alter function by localized lesioning or, more recently, electrical stimulation. The latter has the advantages of being non-permanent, adjustable and reversible. Subthalamic deep brain stimulation for Xxxxxxxxx’x disease is based on the finding that neuronal activity in the STN is abnormal in the Parkinsonian state (14). The degree of improvement as a result of DBS can be predicted by the degree of improvement produced by oral levodopa (15). Globus pallidus deep brain stimulation for dystonia is based on empiric evidence of efficacy (16), although recent evidence of abnormal oscillatory activity in the GPI in dystonia provides some physiologic rationale for the therapy (17,18). DBS for idiopathic primary dystonia is expected to produce at least a 50% improvement In BFMDRS scores (18).
Deep brain stimulation systems consist of an electrode, which is precisely placed in the brain, a subcutaneous extender lead, and an implanted pulse generator (IPG), which is usually placed subclavicularly. The DBS electrodes that are currently commercially available (Medtronic, Minneapolis, MN) have 4 independent electrical contacts that are separated by 1.5 mm or 0.5 mm. The IPG is a programmable unit that can activate any of the contacts in the DBS electrode with varying degrees of electrical stimulation. IPG’s are tuned to provide optimal therapeutic benefit and this may be adjusted on an ongoing basis.
2.2 CONVENTIONAL DBS IMPLANTATIONS: METHODS, EXPERIENCE AND OUTCOMES
The current technical approach for implantation of DBS electrodes involves a complex, highly invasive 6-8 hour procedure performed on awake patients. The primary goal in DBS implantation is to achieve accurate electrode placement (within 1 mm of the desired target) with a minimum of patient risk and operative time.
The standard surgical approach is based on the method of frame-based stereotaxy. In this method, a rigid frame is fixed to the patient’s head, using skull pins, to provide a coordinate system. A “stereotactic” MR or CT scan is then performed that shows both the frame axes and the brain structure. The stereotactic coordinates of the brain target to be implanted, with respect to the frame axes, are calculated and the patient is transported to the operating room. Following scalp incision and creation of a xxxx hole in the skull, instruments are
12
mounted on the frame so as to point through the skull opening to the stereotactic target. Electrode insertion is ultimately achieved with this large externalized frame. After scalp closure, and removal of the stereotactic headframe, the patient returns to the scanner for postoperative verification that the electrode is appropriately placed and to exclude early hemorrhage.
With this surgical approach, the preoperative stereotactic MR or CT images provide the starting point for the procedure. However, conventional stereotaxy using “historical” (preoperative) images does not by itself provide the required accuracy for final DBS electrode placement. In a conventional operating room setting, there is no intraoperative brain imaging technique that has sufficient contrast and resolution to guide and confirm correct electrode placement. As a result, hours are spent performing physiological “mapping” of the brain with multiple penetrations of a microelectrode so as to determine the correct target location based on brain electrical activity in the region of the intended target. This micro-electrode recording (MER) technique is used to map the borders of the target nucleus with greater spatial resolution than is possible with stereotaxy alone (19). The final electrode location may be up to 3 mm away from the initial stereotactic target, based on the correction afforded by intraoperative microelectrode exploration. MER methods are well established for STN DBS in Xxxxxxxxx’x disease, but are much less well established for dystonia. Physiological mapping inherently requires the patient to be awake during surgery and off their usual medications.
2.2.2 UCSF Experience with Conventional DBS Therapy for PD and Dystonia
Drs Starr and Xxxxxx have performed over 900 DBS surgeries at UCSF and the San Francisco Veteran’s Affairs Medical Center since 1998 (3,19-21). While DBS surgery for dystonia has been performed in over 75 patients, the vast majority of DBS surgeries have been performed in patients with moderately advanced PD who had developed motor fluctuations and/or levodopa-induced dyskinesias under optimal medical therapy. These DBS surgeries have been performed with conventional stereotactic methods, including intra-operative micro-electrode recording. Surgical planning is based on a pre-operatively acquired MR data set, which is obtained the morning of surgery with a stereotactic frame mounted to the patient’s skull. In the operative suite, burrhole access is created and MER electrodes are inserted to the preliminary target based on stereotactic localization. Positioning is then refined, if necessary, based on the MER response obtained. This may involve multiple parallel penetrations of the brain with a microelectrode, along parallel trajectories spaced 2-3 mm apart. The DBS electrode is then inserted, test stimulation is performed with assessment of the patient’s responses, and the electrode is then secured to the skull and the skin incision is closed. Surgical time for a bilateral DBS implantation typically takes 6-8 hours with this approach and requires an awake and cooperative patient. The patient is then returned to the MR suite for post-operative documentation of electrode position.
We have investigated the correlation between the MR indicated positions of DBS electrodes within the STN to the achieved clinical outcome in patients with PD (19). Based on our own experience in correlating clinical outcomes and intraoperative physiology with postoperative MR determinations of electrode position in over 800 STN DBS implantations, we have developed the requisite radiographic criteria for successful implantation. For the subthalamic nucleus target, we have found from analysis of postoperative MRI that the clinically effective active contact is located 11-13 mm from the midline, approximately even in the anteroposterior direction with the anterior border of the red nucleus at an axial level 4 mm inferior to the AC-PC plane of the brain. This corresponds to the dorsolateral aspect of the STN and should be a minimum of 2 mm from the internal capsule. If an electrode is placed within this focal MR-visible zone, consistent clinical benefit can be reliably anticipated (19). This suggests that appropriate device localization, as determined by MR imaging, may be sufficient to assure a good clinical outcome for STN DBS in PD. We have performed similar analysis with GPI stimulation for dystonia and found (3) that active electrode locations positioned near the intercommissural plane, a mean distance of 3.6 mm from the pallidocapsular border (or 20 mm lateral, 2.5 mm anterior and 5.8 mm inferior to the midpoint of the AC-PC line) have been associated with the best clinical outcomes.
2.2.3 Outcome Measures Following DBS Therapy
Clinical outcomes and complications associated with conventional surgical practices for DBS implantation, are evaluated by a movement disorder neurologist prior to DBS therapy and followed for prolonged periods post-operatively. [***]
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
13
This study revealed a mean off medication UPDRS-III improvement of 52% with DBS, with a 95% confidence interval of 48-57%. For dystonia, optimal lead placement produced >50% decrease in the BFMDRS score (16).
In terms of complications, asymptomatic hemorrhage was seen in 15 of 637 DBS implantations performed at our site between 1998-2006 (23). This rate of 2.4% is comparable to that previously reported (24) using stereotactic localization supported by MER. Our complication assessment also found poor electrode positioning, revealed by post-operative imaging and requiring re-operation, in 11 of 637 implantations (1.7%). Other complications included infection (1.9%), hemorrhagic stroke (1.3%), post-operative seizures (0.6%) and major air embolus (0.5%). This data provides a relevant retrospective control population against which the outcomes achieved with direct MR guidance can be evaluated.
2.3 PRELIMINARY STUDIES OF MR GUIDED DBS ELECTRODE INSERTION
MR imaging has been performed in the operative setting since the early 1990’s (25.) Inteventional MR has most widely been used to monitor tumor resections, where surgical approach and resection completeness can be monitored intra-operatively. Minimally invasive procedures, such as brain biopsy, have also been demonstrated on closed bore high-field strength MR systems (26), which offer the highest quality MR imaging capabilities. A technique has been proposed to utilize the MR coordinate system as the surgical stereotactic coordinate system. The methodology is referred to as “prospective stereotaxy” (27) and relies on rigid fixation of the head with respect to the bore of the magnet. Real time MR imaging is then used to orient a burrhole mounted trajectory guide towards the intended target. This approach obviates the need for a stereotactic frame or pre-operative scanning and allows both confirmation of procedural success and complication control within the intraoperative session. Furthermore, imaging acquired intraoperatively is not subject to registration errors and permits compensation for dynamic processes such as brain shift.
2.3.1 Methodology for MR Guided DBS Implantation
We utilized the same general principles that had previously been applied to MR guided brain biopsy (28) to develop a prototype system for DBS implantation (29). This approach is performed entirely within the magnet bore and requires a burrhole mounted MR compatible trajectory guide whose orientation can be visualized with MR imaging (Figure 1). An “alignment indicator”, is used to indicate orientation and articulates around a precise point called the “pivot point”. Since the pivot point is fixed, it is important to assure that the burrhole be created in an appropriate location. Thus, an initial contrast enhanced T1-weighted volumetric scan of the brain is performed to reveal cortical surface structure and vessel locations. This data is also used to roughly identify target coordinates based on the relative position of the anterior (AC) and posterior (PC) commissures. A ray is extended from the estimated target position out through the skull, ideally avoiding the lateral ventricle, suici and blood vessels on the cortical surface. The point where this ray exits the skull must then be identified and the burrhole created at this location. We have utilized several methods for identifying this point, including a fluoroscopic MR sequence that is positioned parallel to the skull surface and centered on the ray. The surgeon can probe the skull surface with an MR visible probe while this fluoroscopic sequence is running and identify the desired entry point. The skin is then marked and subsequently the cranial surface is scored at the desired position for the burrhole. For bilateral procedures this will be repeated on the contralateral side.
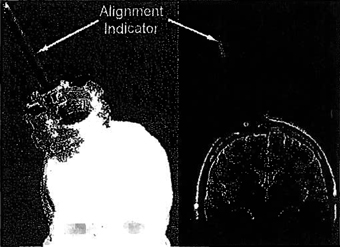
Figure 1: The trajectory guide used in our initial study of MR guided DBS implantations. The plastic burrhole-mounted trajectory guide can be seen on the left with the alignment indicator in place. The corresponding appearance of the alignment indicator on an MR image is shown on the right in an oblique coronal plane.
14
The patient is then moved to the back of the magnet, where a sterile field is established, skin incision is performed, burrholes are created, and trajectory guides are mounted.
The patient is then returned to magnet isocenter, where they will remain until the DBS electrode is appropriately positioned. Scanning is initially performed to locate AC-PC and the mid-sagittal plane. A high resolution T2-weighted turbo spin echo sequence is then acquired in an oblique axial plane that is parallel to AC-PC and perpendicular to the mid-sagittal plane. This standard orientation provides consistent visualization of deep brain structures and permits easy determination of target position with respect to AC-PC. This data set is used to define the deep brain target and it is important to note that it is performed after burrhole creation and dura penetration, which can result in brain shift. The pivot point of the device must also be defined with high precision and this is achieved with two orthogonal MR acquisitions. The pivot point is identified in each data set, the through plane coordinate in each is discarded, and an average is obtained to define the point of articulation of the trajectory guide. The desired trajectory is now set, originating at the deep brain target and extending out through the pivot point of the trajectory guide. A fluoroscopic MR sequence is then prescribed such that it is centered on and perpendicular to this desired trajectory. It is positioned 9-10 cm from the skull surface, such that the pivot point is approximately equidistant from the scan plane and the target point. The trajectory guide is then adjusted until the distal end of the alignment indicator is on the prescribed trajectory (Figure 2).
Once alignment is achieved, the trajectory guide is locked and two orthogonal confirmation scans are performed. The line of intersection of these scans corresponds to the desired trajectory and projections of the alignment indicator are manually made to predict whether the trajectory will intersect the target. Small adjustments in trajectory may be required based on the confirmations scans. At this point the alignment indicator is removed and replaced with a multi-lumen insert (MLI). The MLI has a central channel that is oriented in the same direction as indicated by the alignment indicator. It further has 4 additional parallel channels arranged in a cross pattern that are each offset by 3 mm with respect to the central channel and can be used if initial targeting is substantially inaccurate. Since DBS electrodes are not stiff, a rigid ceramic mandrel within a plastic peal away sheath is initially inserted into the brain. This mandrel is inserted along the prescribed trajectory to the target depth. MR scanning is performed during insertion, to screen for complications and assure the trajectory is being followed, and repeated when the mandrel reaches target depth to assess positional accuracy (Figure 3). If the mandrel position is considered to be sub-optimal, then it will be removed and either re-inserted via a parallel channel of the MLI or a new alignment is performed. Once acceptable positioning is achieved, the procedure can be repeated on the contralateral side if a bilateral implantation is being performed. Following acceptable positioning, the mandrel(s) is removed and the DBS electrode is inserted through the remaining peal away sheath. Another MR scan is performed to confirm
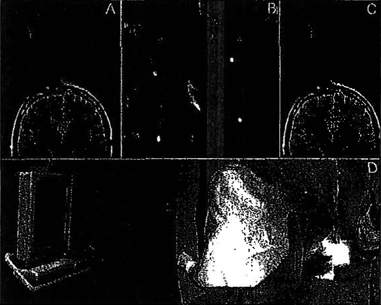
Figure 2: The process of aligning the trajectory guide is summarized. The desired path from the deep brain target to the pivot point is extened out into space and a scan plan is centered on this ray, 9-10 cm from the pivot (A). The surgeon then reaches into the magnet bore and manipulates the trajectory guide while a fluoroscopic MR sequence is run (B) and presented to the surgeon on an in-room monitor (D). B shows four distinct stages of alignment and signal in these frames corresponds to the alignment indicator (bright dot) or the surgeons hand. After alignment the bright dot is centered in this image and the trajectory guide is locked. A confirmation scan is performed (C) after alignment to assure correct orientation.
15
appropriate electrode positioning and the patient is then returned to the rear magnet opening. The DBS electrode is secured and the peal-away sheath is removed, leaving just the electrode in place. Finally, the electrode is secured to the skull, the trajectory guide removed and the skin incision closed. Following conventional practice, the remaining components of the DBS system (pulse generator and lead extender wire) will be placed at a later date in the standard operating room.
2.3.2 Accuracy and Clinical Outcomes
Thirty PD and two dystonia patients have undergone MR guided DBS electrode implantation with our initial prototype. All patients signed an informed consent form that was approved by the university’s committee on human research. Patients either received a single unilateral (n=5) electrode or bilateral (n=27) DBS electrodes. Patient’s receiving bilateral DBS electrodes either had two staged surgical procedures (n=4) or received both electrodes in a single surgical session (n=23).
We report only on PD patients due to the limited number of dystonia studies.
Radiographically acceptable positioning within the STN was achieved on the first pass through the brain in 49 of the 57 (86%) insertions. Two passes through the brain were required in 7 cases (12%) and one case (2%) required three brain penetrations. Mean error from the intended target (Figure 3) on the first pass was 1.2 mm ± 0.7 mm (range = 0.1mm - 2.9 mm). Poor initial positioning requiring mandrel removal and either realignment or use of a parallel channel was associated with relatively large initial errors (2.3 ± 0.5 mm). However, 33% of electrodes with acceptable positioning on the first pass had targeting errors ³ 1.3 mm and may have been refined if the delivery system was capable of achieving such fine adjustments.
Patients who underwent MR guided DBS implantation received neurological evaluation at baseline and 11/25 bilateral implantation patients received follow-up evaluation 6-12 months following surgery. Pre-operative baseline evaluation included evaluation of UPDRS-III, which was performed in the off-medication state as well as in the on-medication state. Post-operative evaluation was performed with optimal DBS stimulation in a similar manner (off and on medication). The degree of improvement as a result of DBS can typically be predicted by the pre-operative degree of improvement produced by the patient’s anti-parkinsonian medications (15,30). Pre-operatively, patients in this study averaged a 61 ± 16% (range: 24-79%) improvement in their UPDRS-III after medications. Baseline off medication UPDRS-III scores improved by an average of 61 ± 28% when compared to the off medication/on stimulation postoperative condition. The range in this outcome was relatively wide (5-89%) but is generally consistent with our own experience of outcomes with electrodes placed in a conventional fashion, published data (31) and degree of pre-operative response to medications (15).
Mean surgical time, measured from skin incision to skin closure, has averaged 220 ±32 minutes over the past 15 bilateral DBS implantations. Unilateral procedures have been performed in as little as 123 minutes and bilateral procedures in as little as 177 minutes. It is important to appreciate that this time includes the target visualization scan, which would be performed pre-operatively with conventional implantations, and all other intra-operative imaging. A final 3D volume, which is actually acquired after skin closure, is also obtained and precludes the need for further post-operative evaluations. Thus, the methodology can be time efficient, compressing pre and post-operative imaging with the surgical procedure in very reasonable surgical durations.
Two early patients developed post-operative wound infections that ultimately required removal of the electrodes, which led to an adjustment in surgical technique and no infections attributable to the DBS electrode have occurred since. One small hemorrhage was acutely detected intra-operatively in a dystonia patient
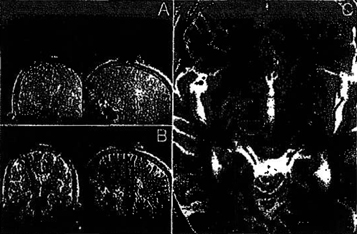
Figure 3: Insertion of the ceramic mandrel is presented. After alignment of the trajectory guide (A), the mandrel is inserted and scanning is performed to monitor insertion and screen for hemorrhage (8). The target selection scan is then repeated (C) to evaluate proximity to selected target (indicated by center of orange circle). The small artifact produced by the mandrel (red arrow) can be more easily appreciated on the contralateral side, where the initial target is not marked.
16
where GPI was targeted. The hemorrhage was monitored to assure that it did not enlarge and proved to be asymptomatic post-operatively.
2.3.3 Limitations of the First Generation Approach
While the initial findings of MR guided DBS implantation have been very positive, there remain substantial technical and clinical limitations that affect the achievable accuracy, precision and likelihood of dissemination to centers with less clinical and technical expertise. These limitations fall into the following categories:
o Trajectory Guide - The present trajectory guide has several key flaws that must be addressed. The physical dimensions of the guide can be problematic when performing bilateral procedures. This frequently required selection of a more lateral burrhole site than desired and occasionally required physical adaptation of the trajectory guide base. The design further requires that the alignment indicator be removed and replaced with an MLI when transitioning between alignment and insertion. Trajectory guide orientation can be affected by this swap and the MLI may not seat correctly when inserted. These limitations were thought to affect several cases where the initial brain penetration was not ideal. The requirement to lean into the bore to adjust the device while monitoring the in-room monitor also proved to be challenging for some surgeons. Finally, the ability to make revisions in position after initial mandrel insertion was very limited. The parallel channels that were offset by 3mm offered only course adjustment and were used in only two leads where the initial accuracy was very poor. The alternative of re-aligning was also not desirable as there was no ability to dial in an appropriate correction. Thus, mandrels whose position was less than ideal were occasionally accepted due to a lack of confidence that substantially improved positioning could be achieved on a subsequent insertion.
o Software Interface - Our initial SW was based on the tools available on the Philips scan prescription and image review platform. Numerous work-arounds and significant attention to detail were required to assure that the desired methodology was realized. For example, determination of the position of potential targets with respect to the mid-point of AC-PC was tedious and time consuming. When exploring potential burrhole sites, it was also difficult to adequately visualize structures that would be intersected on the resulting path to the target. The methodology for finding the selected burrhole site was unnecessarily complicated and may benefit from a surface grid and software to indicate the desired burrhole location on the grid. There were additional factors that potentially affected our accuracy. Specifically, the confirmation scans that were performed after trajectory guide alignment required that we manually define a line running along the alignment indicator and extend it into deep brain structures. Manual definition of this line is prone to small, but relevant, errors and lacks consistency. Confirmation scans performed during mandrel insertion further provided only relatively coarse trajectory information due to the artifact size and the lack of an external alignment indicator during insertion.
o Clinical Validation - Budgetary constraints limited our ability to obtain detailed neurological evaluations prior to and following image guided DBS implantations. This novel methodology makes the acquisition of intra-operative physiologic assessments, which is a staple of convention implantation surgery, largely impractical. Thus, we must be able to clearly demonstrate that DBS electrodes that are appropriately positioned anatomically with intra-operative imaging will consistently produce therapeutic outcomes that are comparable, or superior, to electrodes that are implanted conventionally with supportive intra-operative physiologic evaluations. For widespread acceptance of the image guided approach, it will be necessary to carefully and clearly demonstrate that the absence in intra-operative physiologic data does not compromise the clinical effectiveness of stimulation therapy.
DBS is a rapidly disseminating therapy that would substantially benefit from simplified implantation methodologies. We propose the use of infra-operative MR guidance to deliver DBS electrodes. This approach may obviate the need for physiologic feedback from awake patients, which is currently necessary primarily due to the uncertainty in electrode position with conventional surgical access. If proven to be efficacious, the technique should require shorter surgical periods, fewer brain penetrations, and result in more consistent placement of DBS electrodes than existing surgical methods. While our preliminary results are very encouraging, we have identified numerous areas where the methodology can be improved to both increase accuracy and mitigate the requirement for technical expertise. We have put together a highly skilled set of investigators with the necessary backgrounds for investigating this emerging field and partner with a company ideally positioned to contribute to our stated aims.
17
3. Research Design and Methods
We propose to develop, test and clinically validate a delivery system that is optimized for MR guided DBS implantation. In collaboration with our research partner, Surgi-Vision, Inc, we have identified the limitations of our present approach and defined the requirements for an ideal delivery system. These specifications address many of the issues identified in Section 2.3.3 and should substantially improve our methodology. We initially intend to evaluate the safety and efficacy of the novel delivery platform in phantom models. We will subsequently apply the technique to patients receiving DBS for treatment of Xxxxxxxxx’x disease and dystonia. We will seek clinical validation of the technique, including immediate evaluation of the technical success of the surgical procedure, as well as delayed evaluation of the neurological benefits of DBS.
3.1 PRE-CLINICAL VALIDATION OF THE DELIVERY SYSTEM (Specific Aim 1)
Prototypes of the complete delivery system are anticipated by the proposed start date of this collaboration (October 1, 2009) and will be evaluated as follows:
3.1.1 RF Coil (Milestone 1)
An RF coil will be integrated with the head fixation frame and consist of 9 channels (Figure 4). It will be open anteriorly and superiorly to permit patient access. Five channels will be dedicated to brain imaging and an additional 4 channels will be incorporated into a platform positioned superior to the patient for the purpose of imaging the trajectory guide. Signal to noise analysis (SNR) will be measured with this coil at three key locations and compared to the performance of the existing 2-element array and a conventional 8-channel head coil (In Vivo, Orlando, FL). SNR will be assessed in the center of the coil, corresponding to the approximate location of deep brain structures, and in regions consistent with the location of the pivot point and distal aspect of the trajectory guide. These assessments will be made without applying signal homogeneity corrections and will take the ratio of the mean signal in a region of interest (ROI) at the specified location to the standard deviation (SD) of the noise in a region of the image where no signal is present. Coil performance must exceed that achieved with the 2-element array and approximate the 8-channel head coil in the region of deep brain structures.
[***]
3.1.2 Trajectory Guide (Milestone 2)
A modified trajectory guide has been designed that overcomes many of the limitations identified in Section 2.3.3 (Figure 5). This MR compatible trajectory guide again features a precise point of rotation and an alignment indicator that is visible on MR images. However, it does not require that the alignment indicator be removed prior to mandrel introduction, has a much narrower lateral profile, and can be precisely actuated remotely. Further, it has an additional “X-Y stage” that permits the guide to perform parallel trajectories with precise lateral offsets. Utilization of the X-Y stage requires an understanding of the orientation of the frame with respect to acquired images and so additional MR-visible fiducials are built into its base (arrows). Initial evaluation of the trajectory guide will focus on the detected position of the pivot point, which will be determined with the alignment indicator of the trajectory guide in 25 different orientations with respect to the base. Lateral (device in Figure 5 moves in and out of plane) and longitudinal (device in Figure 5 moves left right) offsets corresponding to angulations of -20°,-10°, 0 , +10°, +20° will be employed with all possible combinations. The detected position of the pivot point based on MR measurements as outlined in Section 2.3.1 will be established for all angulations. The mean and SD of these values will be established to determine if any angulations produce a systematic offset in the pivot point and to determine the consistency in this value

Figure 4: The prototype RF coil being developed for DBS implantation. The coil is integrated with a head frame (black structure) and has elements specifically dedicated to head imaging. Additional elements for imaging the trajectory guide are built into a platform superior to the top of the head (arrow). Also visible are MR visible surface grids, which simplify the procedure for localizing burrhole sites.
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
18
(which should be <0.2mm). Subsequent studies will be aimed at quantifying targeting accuracy and will be performed in collaboration with the developed software. (Section 3.1.3).
3.1.3 Implantation Software (Milestone 1)
A software tool is under development that will break the implantation procedure into the following clearly defined stages : (1) preparation and burrhole localization (2) trajectory guide alignment (3) mandrel insertion and (if necessary) revision (4) completion and report generation. The software tool will be run on an independent workstation with a direct network connection to the MR acquisition console. This approach has the advantage of being independent to the MR system manufacturer and specific MR software release. The objective of the software is to intuitively lead the clinician through the implantation procedure, present optimal visualization of the procedure, and to provide guidance with regard to the required geometric properties of MR acquisitions. Initial software evaluations will be performed with specific SW components, with full evaluation subsequently performed in Section 3.1.4. There are several novel components that must be validated, including automated AC, PC and mid-sagittal plan detection, alignment indicator projection and X-Y stage offset settings. Automated AC, PC and mid-sagittal plan detection will be performed on pre-operative data obtained in 10 DBS patients. Two trained neurosurgeons (PS, PL) will independently identify the spatial coordinates of AC and PC and the angulation of the mid-sagittal plane. Correlation within and between the neurosurgical practitioners and the SW will be established. Next, projection of the alignment indicator will be tested in a saline filled phantom. The trajectory guide will be positioned in the same 25 orientations indicated in Section 3.1.2, MR scans will be acquired and automatic projections created. A rigid ceramic mandrel will then be inserted through the alignment indicator to provide truth, and the difference between the mandrel position and ray prediction 8 cm (~ target depth) below the pivot point quantified. The results will be interrogated for systematic errors and mean relative errors <0.5mm will be sought. Finally, the X-Y stage offset will be tested by deliberately misaligning the trajectory guide to be 1-3 mm from the intended target. The desired offset vector will be specified and the necessary translation of the X-Y stage calculated based on the relative orientation and angulation of the trajectory guide to the scan plane in which the target was identified. The mandrel will then be withdrawn, the X-Y stage translated and the mandrel re-inserted. The magnitude and direction of any residual difference between the intended and actual mandrel location will be quantified to determine revised targeting accuracy and exclude systematic errors. [***]
3.1.4 Phantom Assessment of System Accuracy (Milestone 2,3)
The complete delivery system will be tested in a head-mimicking fluid-filled phantom containing a series of distinct slots. A trajectory guide will be rigidly mounted to the phantom such that its base is angled at -20°, 0°, or +20° with respect to the plane of the target. Alignment and mandrel insertion will be performed for 8 separate targets at each base angulation, and this experiment will be performed on at least 2 separate days to establish targeting accuracy, consistency and potential for systematic errors. Alignment of the trajectory guide will follow the methodologies outlined in detail in Section 3.2.2 and accuracy will be assessed by the vector difference between the target coordinate and mandrel position at target depth. [***]
3.2 MR GUIDED DBS IMPLANTATION IN XXXXXXXXX’X DISEASE (Specific Aims 2, 4)
This single center pilot study will evaluate MR guided DBS implantation in the STN of Parkinsonian patients.
3.2.1 Patient Recruitment
Twelve adult patients with PD will initially be recruited during months 3-9 (Milestone 4,5) of this proposal (statistical justification in section 3.5). Pending acceptable initial findings, an additional 18 patients will be

Figure 5: The new trajectory guide is shown. It includes a base with MR fiducials (arrow) a vertical alignment indicator and articulation knobs that can be precisely manipulated remotely.
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
19
recruited (statistical justification in Section 3.5). Patients will be recruited from the UCSF surgical movement disorders clinic referred for DBS surgery for PD. Our center is the busiest DBS surgical center on the West coast and currently evaluates over 75 new patients a year for DBS candidacy. Patients with disabling symptoms of advanced PD that are inadequately controlled with medical therapy will be considered for the study. Inclusion/exclusion criteria will be identical to the criteria for patients undergoing surgical treatment for PD by standard methods. Inclusions are: (1) diagnosis of bilateral, idiopathic PD with clinically significant motor fluctuations despite maximized antiparkinsonian therapy (2) at least 5 years’ duration, relative to the date of surgery, since diagnosis of PD, (3) age >21 inclusive, on date of surgery, (4) the subject is ambulatory in their best on time (not wheelchair bound), (5) the subject is medically able to undergo the surgery as determined by clinical and laboratory evaluations (e.g., subjects will have normal coagulation tests and normal platelet levels), and (6) the subject is expected to be able to comply with and understand the required visit schedule and all required tests and procedures. Exclusions will include: (1) a history of any clinically significant medical, psychiatric, or laboratory abnormality for which participation in the study would, in the opinion of a mental health expert (CR), pose a safety risk to the subject, (2) history of treatment of PD by any procedure involving intracranial surgery or implantation of a device, (3) MR of the brain within 12 months before the surgery which demonstrates an intracranial abnormality that would contraindicate surgery (e.g. stroke, tumor, vascular abnormality affecting the DBS target area), (4) any disorder that precludes a surgical procedure (e.g., signs of sepsis or inadequately treated infection) or alters wound healing, (5) receipt of antiplatelet agents for at least 10 days prior surgery, (6) a score of less than or equal to 24 on the Folstein Mini-Mental examination performed during the eligibility evaluation period, (7) history of significant psychiatric illness, epilepsy, or Alzheimer’s disease, (8) active drug or alcohol abuse, (9) pregnancy or lack of effective contraception in women of childbearing potential defined as one year post-menopausal or surgically sterile, (10) treatment with nonantiparkinsonian agents (e.g., typical neuroleptics) that may affect symptoms of PD within 60 days before entering the study, (11) any medical disability (e.g., severe degenerative arthritis, compromised nutritional state, severe peripheral neuropathy) that would interfere with the assessment of safety and efficacy in this trial, and (12) inability to follow-up with post-operative study visits. Written consent will be obtained prior to the surgical procedure and the investigators will extensively discuss with patients and their families the study rationale, protocol, risks, and potential benefits.
3.2.2 Surgical Procedure
Each patient will undergo bilateral STN-DBS implantation, which will take place entirely within the MR suite. Implantation will follow the general methodology outlined in Section 2.3.1, but will be performed with the delivery system validated in Section 3.1. The patient will be anesthetized and immobilized in the RF coil. Surface grids will be placed bilaterally on the skull and the patient moved into the magnet bore. MR contrast (Magnevist, Bayer HealthCare) will be administered and a volumetric T1-weighted MR scan will be performed to reveal brain structure and vessel location. Standard offsets for the STN target (3mm posterior, 12mm lateral and 4 mm inferior to mid-point of AC-PC) will be assumed and possible trajectories will be explored. Once an acceptable trajectory is identified, the exit coordinates on the external grid will be identified and a xxxx will be made on the skull with a punch tool. This procedure will be repeated for the contralateral side. The patient will then be moved to the rear magnet opening and a sterile field established. Skin incisions and burrholes will be created at the marked sites and the exposed dura mater opened. Finally, the trajectory guides will be mounted and the remote actuator systems attached.
The patient will be returned to magnet isocenter and high quality T2-weighted images will be acquired in an oblique axial plane parallel to AC-PC. Bilateral targets within the dorsolateral STN will be identified in this dataset by an attending neurosurgeon (PS or PL). MR scanning will additionally be performed on the trajectory guides to identify the point around which they articulate (pivot point), the orientation of the base with respect to the patient’s anatomy, and the initial orientation of the alignment indicator. Trajectory guide alignment and mandrel insertion will be performed serially, with acceptable mandrel positioning achieved on one side before continuing to the contralateral side. Fluoroscopic MR imaging through the distal aspect of the trajectory guide will be run while the surgeon remotely manipulates the trajectory guide. Real-time feedback showing an automatically generated ray projection from the trajectory guide onto the image slice that the target was identified on will be presented to the surgeon on the in-room monitor. Once this projection intersects the identified target, the fluoroscopic sequence will be interrupted and two orthogonal MR images along the desired trajectory acquired. The orientation of the alignment indicator will be evident in these images and its
20
orientation automatically detected. A projection of this orientation will be extended to target depth and it will be determined whether the present alignment adequately intersects the selected target. If it is unacceptable, an adjustment will be defined, a trajectory guide correction executed, and the confirmation scans re-acquired.
After acceptable alignment, a ceramic mandrel within a plastic peel-away sheath will be introduced via the central channel in the alignment indicator. Several (typically 2-3) confirmation scans will be obtained as the mandrel is slowly advanced to target depth. This insertion will be aborted if an unacceptable offset is detected, but otherwise will continue to target depth. At this point the procedure will be repeated on the contratateral side until both mandrels are positioned at target depth. The high quality T2-weighted MR data set that was used to identify the targets will be re-acquired and the locations of the mandrels will be compared to the desired coordinates to determine targeting accuracy (Specific Aim 2). If either mandrel’s position is unsatisfactory, then an offset vector in the scan plane showing the target will be defined. The mandrel requiring adjustment will be removed. Based on the relative orientation of the trajectory guide base and this scan plane, an offset to the X-Y stage of the trajectory guide will be prescribed such that it should produce a parallel trajectory that intersects the intended target. The mandrel will then be re-inserted to the target depth and the new targeting accuracy will be assessed to determine the success of this revision (Specific Aim 2). If necessary, this process will be repeated until acceptable positioning is achieved.
After appropriate positioning, the ceramic mandrels will be removed, leaving the hollow peel-away sheaths terminating and the desired deep brain location. DBS electrodes are then inserted via this channel to the specified depth and a confirmation scan is obtained. The patient is then returned to the rear magnet opening, where the DBS electrode is secured to the skull and the trajectory guide is removed. A final volumetric T1-weighted MR scan will be acquired to document electrode contact positions and orientations.
Following implantation, patients will be hospitalized for 1 or 2 days postoperatively in a step-down unit, as is the case for patients undergoing DBS outside of this protocol. Within 2 weeks following DBS electrode implantation, patients will undergo implantation of the rest of the DBS system (pulse generator, placed subcutaneously in the chest, and extension wire from brain electrode to pulse generator) in a regular operating room. The methods for pulse generator and extension wire placement are identical to those used for patients undergoing conventional DBS surgery.
3.2.3 Data Collection
Technical efficacy of the implantation procedure will be assessed based on the following parameters: (1) the difference, on the first pass, between the selected target position and actual ceramic mandrel position in the axial scan plane used for target selection (2) the final difference between the selected target position and actual ceramic mandrel after any revision (3) mean distance between the selected target and active contact of the DBS electrode, both determined in AC-PC coordinates (4) mean number of brain penetrations required to produce acceptable electrode position and (5) surgery duration as measured from skin incision to skin closure. We will further track surgical complications including symptomatic and asymptomatic hemorrhage and infection. The study will be deemed a failure if any of the following occur: (a) initial targeting accuracies that exceed a radial error of 2mm in more than 50% of implantations, (b) a final mean radial error >1.5mm, (c) mean number of brain penetrations >2 and (d) hemorrhage or infection rates that are deemed clinically unacceptable. These parameters will be explicitly interrogated after completion of the first 12 PD patients (Milestone 6) to justify study extension (Milestone 8) and continuously monitored throughout the study.
3.3 MR GUIDED DBS IMPLANTATION IN DYSTONIA (Specific Aim 3)
This single center pilot study will evaluate MR guided DBS Implantation in the GPI of dystonia patients. GPI represents a distinct deep brain target at a depth comparable to the STN.
3.3.1 Patient Recruitment
A total of 12 patients will be recruited from the UCSF surgical movement disorders clinic referred for DBS surgery for dystonia (Milestone 9) statistical justification in section 3.5). Our busy DBS surgical center currently evaluates over 20 new dystonia patients a year for DBS candidacy. The following inclusion criteria will be applied: (1) Severe idiopathic or secondary dystonia diagnosed by a movement disorders neurologist (JO), (2) severe functional impairment despite optimal medical management, including failed botulinum toxin therapy if appropriate, (3) age >7 inclusive, on date of surgery, (4) the subject is medically able to undergo the surgery as determined by clinical and laboratory evaluations, and (5) the subject is able to comply with and
21
understand the required visit schedule and all required tests and procedures. Exclusions will include: (1) medical contraindications to surgery, stimulation, or magnetic resonance imaging, (2) active alcohol or drug abuse, (3) pregnancy and (4) Inability to follow-up with post-operative study visits. Written consent will be obtained prior to the surgical procedure and the investigators will extensively discuss with patients and their families the study rationale, protocol, risks, and potential benefits.
3.3.2 Surgical Procedure and Data Collection
Each patient will undergo unilateral or bilateral GPI-DBS implantation, which will take place entirely within the MR suite, with the surgical approach detailed in Section 3.2.2. In this case, standard offsets for GPI will be used during trajectory planning (20mm lateral, 3 mm anterior to mid-point of AC-PC) and targets within the GPI will be identified on inversion recovery (IR-TSE) MR images. Post-operative procedures, including implantation of the remaining DBS components, will follow those outlined in Section 3.2.2 and match those used for dystonia patients who received DBS electrodes in conventional fashions. Technical efficacy will be evaluated with the same measures outlined In Section 3.2.3, with mandrel position error evaluated in the oblique axial plane in which the GPI target was identified (typically AC-PC plane). Conditions for study failure will remain the same, although hemorrhage risk in GPI is known to be higher.
3.4 EVALUATION OF CLINICAL EFFICACY (Specific Aim 4)
Technical efficacy of this procedure will be evaluated in Sections 3.2 and 3.3. Neurological and neuropsychological testing will also be performed on all study participants to establish clinical benefit. Each subject will undergo a pre-surgical screening period of up to 1 month to establish their baseline state and study eligibility, and post-surgical evaluations will occur 6-12 months post-implantation.
3.4.1 PD Neurological Evaluations
Patient’s will undergo a comprehensive neurological evaluation pre-operatively and again at 6-12 months following surgery (Milestone 4,7,8). The evaluation will be performed by a board certified neurologist who specializes in movement disorders (JO). Clinical efficacy of bilateral STN DBS will be assessed using multiple outcome measures. Clinical outcome will primarily be assessed based on the degree of benefit from baseline off medication UPDRS III motor score to postoperative off medication scores with stimulation on. The predetermined criteria for study failure will be a mean off medication UPDRS-III improvement with stimulation on of less than 40%. Additional secondary clinical outcome measures will be analyzed and include change from baseline to postoperative state in the following criteria: (1) individual UPDRS I (Mentation, Behavior and Mood), II (Activities of Daily Living), III (Motor, off medication/on stimulation), and IV (Complications of Dopaminergic Therapy) scores, (2) total UPDRS score (3) Stand-Walk-Sit Timed Motor Test results (4) Dyskinesia Rating Scale score (5) patient motor diary recorded values of the percent on time without dyskinesia, on time with mild dyskinesia, on time with severe dyskinesia, and off time (6) PD Questionnaire-39 (PDQ-39 - a quality of life PD specific instrument) score, (7) SF-36 questionnaire (a commonly used health survey), and (8) investigator- and subject-rated clinical global impressions (CGI) score. Percent reduction in concomitant antiparkinsonian medications after surgery, ideal DBS electrical settings, and active electrode location as determined by post-implantation MR imaging will also be determined and analyzed.
3.4.2 Dystonia Neurological Evaluations
While the study is not powered to provide a statistically significant evaluation of clinical efficacy in dystonia, we will assess the clinical benefits that are realized in this patient group. Patient’s will undergo a comprehensive neurological evaluation pre-operatively and again 6-12 months following surgery (Milestone 9,10). The evaluation will be performed by a board certified neurologist who specializes in movement disorders (JO). The primary clinical efficacy of bilateral GPI DBS will be assessed by the percent change from baseline in the BFMDRS movement subscore to the 12 month postoperative subscore Additional secondary outcome measures will include: (1) BFMDRS disability subscale (2) TWSTRS (Toronto Western Spasmodic Torticollis) severity, disability, pain, and total score (3) other cerebral palsy motor rating scales if appropriate (4) SF-36 questionnaire and (5) investigator- and subject-rated CGI score. Percent reduction in concomitant anti-dystonic medications, the ideal DBS electrical settings, and the active electrode location as determined by post-implantation MR imaging will also be determined and analyzed.
22
3.5 STATISTICAL JUSTIFICATION FOR SAMPLE SIZES
The justification for the sample sizes proposed in this study is based on both targeting accuracy (technical efficacy) and clinical outcomes based on neurological evaluations (clinical efficacy). Targeting accuracy is assessed based on our ability to localize a selected three-dimensional target. In our preliminary series of patients with our initial prototype we documented a targeting error of 1.2±0.7 mm. We anticipate improving the accuracy and consistency of these values and estimated our new targeting error to be s1.0±0.5 mm. With this assumption, a sample site of 12 patients (24 electrodes) is sufficient to reduce the 95% confidence interval of the standard error of the mean to <0.2mm. This sample size should therefore be capable of demonstrating targeting accuracy that is better than or equal to that achieved with the initial prototype. We will initially validate the accuracy of our new delivery system on PD patients in which the STN is targeted (Specific Aim 2). If this aim is met, we will then recruit 12 patients (24 electrodes) with dystonia and determine targeting accuracy in a deep brain structure (GPI)(Specific Aim 3).
For PD patients we propose a total of 30 patients, which is based on our ability to demonstrate equivalence to the therapeutic benefits afforded by medication prior to surgery (Specific Aim 4). This is the same standard to which electrodes implanted with conventional methodologies are held (15). We assumed a standard deviation in UPDRS-III values of 10, which is based on average on-medication UPDRS scores of 19±8 on medication and 17±14 on stimulation in our preliminary series. We performed a power analysis and determined that 27 patients would be necessary to be able to detect a difference in UPDRS outcomes with medication versus stimulation of >8 UPDRS units (power = 0.8). We allow for 10% of patients lost to follow-up, producing the proposed sample size of 30 patients.
3.6 DATA ANALYSIS (Milestone 11)
Data related to technical efficacy will be evaluated to determine the mean, standard deviation and range of the error between the selected target and eventual mandrel position on the first, and potentially subsequent, passes into the brain. We will also calculate the vector difference between the AC-PC coordinates of the selected target and the AC-PC coordinates of the DBS electrode location on the same axial plane as that used for target selection. The first measure provides the most direct measurement of technical success of the device insertion method, while the second provides an overall “application accuracy” of this technique using the actual final location of the DBS electrode. Finally, the mean, standard deviation, and range of the number of brain penetrations and procedure duration will be determined. These results will address Specific Aims 1-3 of this proposal.
Therapeutic outcomes of stimulation therapy in PD will be correlated with the degree of improvement produced by anti-parkinsonian medications pre-operatively. The latter represents the best available standard for predicting the therapeutic benefit that stimulation therapy should achieve in each individual (15,30). A paired t-test and/or Wilcoxin signed-rank test will be applied to the data to determine the correlation between these two independent measures. The other parameters discussed in Section 3.4.1 will also be analyzed for differences between off medication baseline and off medication/on stimulation post-operative states. Significant differences in any outcome variable between the two groups will be assessed with logistical regression using commercially available software (such as Statview V, SAS Institute, Cary, NC). These results will address Specific Aim 4 of this proposal.
There is considerable variability in the clinical characteristics of dystonia, so patient evaluations will be grouped according to their presentation. These groupings are anticipated to include juvenile onset idiopathic dystonia, positive for the DYT1 mutation; juvenile onset idiopathic dystonia, negative for the DYT1 mutation; adult onset craniocervical idiopathic dystonia; tardive dystonia; and secondary dystonia. The results of this study will be compared to outcomes achieved in a prior study within our institution (3). Significant differences in any outcome variable between the two groups will be assessed with logistical regression using commercially available software.
For PD patients, additional assessment will be made of the correlation between implantation accuracy (Specific Aim 2) and clinical outcomes (Specific Aim 4). Targeting accuracy will, in general, vary between hemispheres. Thus, we will correlate stimulation induced improvement in the lateralized limb subscores of the UPDRS-III to the targeting error for the contralateral implant. Xxxxxxx’x product-moment correlation analysis will be performed on these two independent measures to determine how strongly they are related.
23
[See Attached]
24
Budget and In-kind Justification and Facilities (DRT)
1. Budget Justification
[***]
2. In-kind Justification
[***]
3. Facilities and Resources.
[***]
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
25
Exhibit C
In-Kind Contributions
[***]
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
26
ADDENDUM TO SPONSORED RESEARCH AGREEMENT
This Addendum to Sponsored Research Agreement (this “Addendum”) is made effective as of February 4 th, 2010 by and between SurgiVision, Inc., a Delaware corporation (“SVI”) and The Regents of the University of California on behalf of its San Francisco campus (“UCSF”).
A. SVI and UCSF entered into a Sponsored Research Agreement in August 2007, as amended pursuant that certain First Amendment to Sponsored Research Agreement made effective as of December I, 2008, and that certain Second Amendment to Sponsored Research Agreement made effective as of May I, 2009, and that certain Third Amendment to Sponsored Research Agreement made effective as of November 2, 2009 (as amended, the “Research Agreement”).
B. SVI and UCSF wish to supplement the terms of the Research Agreement as set forth below to address certain additional research activities.
NOW, THEREFORE, for good and valuable consideration, the sufficiency and receipt of which are hereby acknowledged, it is hereby agreed as follows:
1. Construction and Interpretation. The provisions of this Addendum supplement, and in no way replace or supersede, the provisions of the Research Agreement. Without limiting the generality of the foregoing, the research activities and funding set forth in this Addendum (including the Exhibits hereto) are in addition to, and do not in any way modify or replace, the research activities, funding and other support set forth in the Third Amendment to Sponsored Research Agreement with respect to the research proposal entitled “Optimized Methodology for Implantation of DBS Electrodes”, for which UCSF received UC Discovery Grant support.
2. Additional Research Activities. UCSF will perform the research activities described in the Research Project Scope of Work attached hereto as Exhibit A (the “Additional SOW”).
3. SVI Support for Additional Research Activities. SVI will provide to UCSF additional funding in an aggregate amount up to $[***] (the “Additional Funding”). UCSF will allocate and apply the Additional Funding to carry out the research activities as described in the Additional SOW. SVI will pay to UCSF the Additional Funding in a single installment within thirty (30) days of the effective date of this Addendum. For the avoidance of any doubt, this Addendum does not affect SVI’s funding obligations set forth in the Third Amendment to Sponsored Research Agreement.
IN WITNESS WHEREOF, SVI and UCSF have entered into this Addendum to be effective as of the date first set forth above.
| THE REGENTS OF THE UNIVERSITY OF CALIFORNIA |
SURGIVISION, INC. | |||||||
| By: | /s/ Xxx Xxxxxxxx | By: | /s/ Xxxxx Xxxxxx | |||||
| Name: | Xxx Xxxxxxxx | Name: | Xxxxx Xxxxxx | |||||
| Title: | Industry Contracts Manager Office of Sponsored Research University of California San Francisco |
Title: | Vice President, Business Affairs | |||||
| Date: | 2/4/10 | Date: | February 4, 2010 | |||||
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
27
Exhibit A to Addendum to Sponsored Research Agreement
Research Project Statement of Work:
TITLE: A Comparison of Navigus II MR (NeXframe MR) and ClearPoint for Workflow and Accuracy in a Cadaver Model
GOAL: Perform procedures using the NeXframe MR and the ClearPoint System to access targets in a cadaver head. Comparison data will be obtained to assess: (1) the overall workflow for the two devices as assessed by the time necessary to complete the respective procedures; and (2) the relative accuracy of the two devices, as assessed by comparing the final tip position with the intended position. The procedures will be performed per the labeling of the two devices.
TEST METHODS / PROCEDURE
[***]
BUDGET
[***]
| [***] | Indicates portions of this exhibit that have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
28