FIRST ADDENDUM TO THE RESEARCH AND LICENSE AGREEMENT
Exhibit 10.5
FIRST ADDENDUM
TO THE RESEARCH AND LICENSE AGREEMENT
This First Addendum to Research and License Agreement (the “First Addendum”) is made by and between The Technion Research & Development Foundation Ltd. (“TRDF”) and Eloxx Pharmaceuticals Ltd. (“Licensee” or “Eloxx”).
WHEREAS, TRDF and Eloxx are parties to Research and License Agreement with an effective date of August 29th, 2013, as amended on November 26, 2013, January 14, 2014 and June 9, 2014 (the “Agreement”); and
WHEREAS, the parties desire to continue the relationship contemplated by the Agreement and therefore to further amend the Agreement as set forth herein;
NOW, THEREFORE, the parties hereby agree as follows:
| 1. | Unless otherwise defined herein, capitalized terms used in this First Addendum shall have the meanings assigned thereto in the Agreement. |
| 2. | The Parties wish to add new specific research work under this First Addendum, all as described in Exhibit D1 attached hereto which shall be added to Exhibit D of the Agreement (the “First Addendum Research”). |
| 3. | For the First Addendum Research, a separated budget is required to be paid by Eloxx to TRDF, all as agreed upon and set in Exhibit D1, in the total amount of thirty thousand US dollars ($30,000) (the “First Addendum Budget”). |
| 3.1 | Licensee shall fund the First Addendum Budget to be performed within 3 months commencing as of the execution of this First Addendum, as follows: |
| 3.1.1 | First installment of ten thousand US dollars ($10,000) shall be paid upon the execution of this First Addendum. |
| 3.1.2 | Second installment of ten thousand US dollars ($10,000) shall be paid no later than June 30th, 2014. |
| 3.1.3 | Third installment of ten thousand US dollars ($10,000) shall be paid upon completion of the First Addendum Research and supply to Eloxx of related materials. |
| 3.2 | VAT as applicable on time of payment, shall be added to each installment. |
| 3.3 | TRDF shall issue a proper invoice for each installment. |
| 4. | Except as added herein, all Confidential terms and conditions of the Agreement shall remain in full force and effect, as relevant to the First Addendum Research. |
1
| 5. | This First Addendum may be executed in counterparts, each of which shall be deemed an original and all of which together shall constitute one and the same instrument. Any signature page delivered by facsimile or electronic image transmission shall be binding to the same extent as an original signature page. |
IN WITNESS WHEREOF, the parties hereby accept and agree to the terms and conditions of this First Addendum.
| ELOXX PHARMACEUTICALS LTD. | THE TECHNION RESEARCH & DEVELOPMENT FOUNDATION LTD. |
|||||||||
| By: | /s/ Xxxxxx Xxxxxx |
By: | /s/ Xxxxxxxx Xxxxxx |
|||||||
| Name: Xxxxxx Xxxxxx | Name: Xxxxxxxx Xxxxxx | |||||||||
| Title: CEO | Title: Technology Transfer Office, Manager | |||||||||
| Date: 3/8/14 | Date: 07.14.2014 | |||||||||
Exhibit D-1
Submitted to Eloxx Pharmaceuticals LTD
Research Plan
Title
Development of Aminoglycose-Based Drug for Treatment of Human Genetic Diseases and Many Forms of Cancer Caused by Nonsense Mutations
I. Scientific and Technological Background
Nonsense mutations are in-frame premature termination codons (PTCs) that convert a sense codon of mRNA to UAA, UAG or UGA stop codon and lead to the production of truncated, nonfunctional proteins. PTCs are responsible for more than 1,800 inherited human diseases, including cystic fibrosis (CF), Duchenne muscular dystrophy (DMD), Xxxxx syndrome (USH), Hurler syndrome (HS) and numerous types of cancer. For many of those diseases there is presently no effective treatment and the only treatment widely used is symptomatic.
One potential approach to treatment considers the use of small molecule drugs to selectively suppress the normal proofreading function at PTCs, but not at normal termination codons. This leads to a favorable competition of near-cognate aminoacyl-tRNAs with the release factor and to the insertion of a near-cognate amino acid at PTCs, allowing continued translation to full-length proteins. This approach, also called “translational readthrough” or “suppression therapy”, was first validated by using aminoglycoside (AG) antibiotics. Numerous in vitro and in vivo experiments including clinical trials have demonstrated the ability of selected structures of AGs (namely gentamicin, paromomycin and G418, Fig. 1) to induce readthrough at PTCs and partially restore functional proteins. However, severe side-effects of AGs, including high human toxicity, along with the reduced readthrough efficiency at subtoxic doses, have limited their clinical benefit for suppression therapy.
AGs selectively bind to the decoding A site on the 16S subunit of bacterial rRNA, and kill bacteria by disturbing the fidelity of the decoding process. Although prokaryotic selectivity is critical to their utility as antibiotics, they are not perfectly selective for the bacterial ribosome; they also bind to the eukaryotic A site resulting in PTC readthrough. Gentamicin and paromomycin are three orders of magnitude more selective to the prokaryotic versus the eukaryotic ribosome. For suppression therapy, this necessitates their use in high quantity, which in turn causes deleterious toxic side-effects, and hence, largely limits their utility.
A noteworthy exception is G418. In addition to its strong antibacterial activity, it also exhibits the highest readthrough activity among all AGs tested to date. G418 is however very cytotoxic to mammalian cells. It has not been clear whether its high cytotoxicity is due to higher specificity to the mammalian ribosome or to some other feature. Clearly, a systematic search for new structures with improved PTC suppression activity and lower toxicity, along with a deeper understanding on structure-activity-toxicity relationship, are required to extrapolate the approach to the point where it can actually help patients suffering from genetic diseases caused by nonsense mutations.

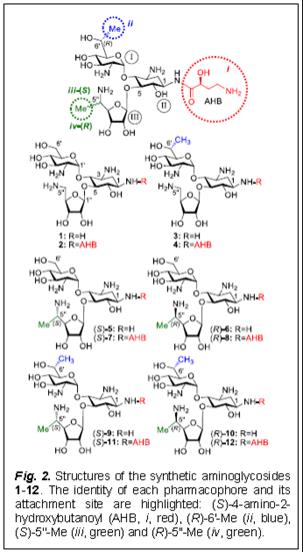
Towards these ends, we hypothesized that by separating the structural elements of AGs that induce readthrough from those that affect toxicity we might obtain potent AG-derivatives with improved readthrough activity and reduced toxicity. By systematically fine-tuning the structure-activity-toxicity relationship, we recently reported a series of structures, 1-8 (Fig. 2), exhibiting significantly reduced toxicity and higher PTC suppression activity than either gentamicin or paromomycin. Protein translation inhibition studies along with antibacterial tests indicated that 1-8 have increased selectivity in their action towards eukaryotic cells than towards prokaryotic cells in comparison to gentamicin and paromomycin. However, none of those leads were able to outreach G418’s peak suppression potency, nor its elevated eukaryotic specificity.
Timor Baasov
The observed increased selectivity of action of 1-8 towards eukaryotic versus prokaryotic ribosome along with their reduced toxicity drew our attention and prompted us to ask several fundamental questions: what structural and mechanistic features are responsible for the observed selectivity increase and toxicity decrease of these synthetic derivatives? Can a general molecular principle for their structure-activity-toxicity relationship be devised? Using this principle, can a synthetic variant with similar or higher PTC suppression activity and lowered toxicity than those of G418 be generated?
To address these questions, very recently we reported (see reference in J. Med. Chem. 2012, in the publications list of the PI) on the design, synthesis and evaluation of a new set of structures, 9-12 (Fig. 2) that perform better than G418 by the above criteria while exhibiting lower toxicity. Furthermore, by using a series of comparative readthrough, protein translation inhibition, antibacterial and toxicity assays between standard and the entire set of designer aminoglycosides 1-12, we demonstrated that the increased specificity towards human cytoplasmic ribosome correlates with the increased PTC suppression activity, and that the decreased specificity towards mitochondrial ribosome confers, at least in part, to the lowered cell toxicity. These observations provide proof of principle that antibacterial activity and toxicity of aminoglycosides can be dissected from their suppression activity. The data further indicated that AG-induced inhibition of cytoplasmic ribosome is a key determinant for PTC suppression activity, and that the inhibition of mitochondrial ribosome is key to AG-induced cell toxicity. These results are therefore beneficial for further research on the development of AG-based drug for the treatment of genetic diseases caused by nonsense mutations.
2. Market Survey
The National Institutes of Heanth (NIH) Office of Rare Diseases estimates that genetic disorders are responsible for the majority of rare diseases and that these diseases affect 25 million people in the US. Similar number of people is also affected in European Union, (EU, estimated 29 million). Orphan diseases are rare and often debilitating conditions, defined in the European Union (EU) as having a prevalence of no more than five per 10,000 people. There are between 5,000 to 8,000 different rare diseases. It is estimated that, on average, 5-15% of patients with any of at least 1,800 distinct genetic disorders have a nonsense mutation as an underlying cause of the disease. Orphan drugs are those medicines used in the diagnosis, prevention or treatment of orphan diseases.
Orphan drugs are a growing issue of importance to American and European healthcare policy makers. The success of orphan drug legislation has resulted in an increasing number of licensed medicines for rare diseases, and many more yet unlicensed products have received orphan drug designation. Several studies estimate a steady increase during 2010-2020 years in the cumulative number of diseases for which an orphan drug is approved (Fig. 3), averaging just over 5 new diseases per year over the next 10 years. The annual per patient cost of existing orphan drugs was seen to vary between €1,251 and €407,631, with the median cost
5
Timor Baasov
being €32,242 per year. The share of the total pharmaceutical market represented by orphan drugs is predicted to increase from 3.3% in 2010 to a peak of 4.6% in 2016 after which it is expected to level off through 2020, as growth falls into line with that in the wider pharmaceutical market.
Since to date CF is the most studied disease in this direction, we will focus on this disorder in regard to the market of the potential drugs. Currently, more than 1,000 different CF-causing mutations in the CFTR gene were identified, and 5-10% of the mutations are premature stop codons. In Ashkenazi Jews, the W1282X mutation and other nonsense mutations account for 64% of all CFTR mutant alleles. In the CF pipeline, there are promising new therapies designed to rectify the cause of CF—a faulty gene and/or its faulty protein product. Bellow is a “snapshot” of those potential CF therapies that are currently in development.
GENE THERAPHY: Because a faulty gene causes CF, adding normal copies of the gene to cells should correct these cells and ultimately cure the disease. Copernicus Therapeutics, Inc. developed an approach that delivers normal copies of the CF gene as tiny particles that slip into CF cells (the development is currently at Phase 2).
PROTEIN ASSIST/REPAIR: This therapy is designed to correct the function of the defective CFTR protein made by the CF gene to allow chloride and sodium to move properly in cells lining in the lungs and other organs. This therapy is not affected by the delivery challenges that have limited gene therapy and enzyme replacement therapy. In addition, it does not necessitate the delivery of foreign genetic material or viruses that gene therapy requires. It is anticipated that by addressing the underlying cause of the disease, small molecule drug might decrease dependence on palliative interventions and ameliorate the debilitation and mortality in patients with genetic disorders. In this category the following potential drugs are in pipeline:
a) PTC124 (PTC Therapeutics, Inc.) – The new drug candidate, PTC124, is designed to repair one type of CF gene mutation, namely nonsense mutation that causes the CFTR protein to stop being made in the cell before it is complete. PTC124 has been on 2012 in phase 3 trial in CF and in DMD patients. These preliminary results in patients with CF and DMD provide confirmation of proof-of-concept that the compound that was originally designed by this company to suppress premature stop mutations (PTC124) can indeed induce ribosomal readthrough of nonsense mutations as an approach to treat genetic disorders.

b) VX-770 (Vertex Pharmaceuticals, Inc.) – This new compound is called a “potentiator” and it may act upon the CFTR protein and help to open the chloride channel in CF cells. Phase 3 dosing has been completed in patients, and since November 2012 the compound is approved by FDA as a drug.
NBs vs PTC124: PTC124 also named as Ataluren (PTC Therapeutics, Inc. USA) is the most advanced drug candidate investigated today for the treatment of genetic defects resulting from nonsense mutations (Phase 3 in CF and DMD). However, yet several problems are faced with the use of Ataluren for the suppression therapy: (1) no real phenotype improvements have been seen after the clinical trials in patients of both diseases; (2) the mechanism of its action is still not conclusive; (3) its action as a readthrough inducer is somewhat controversial since several labs reported its lack of action in different diseases models while others reported opposite data.
In contrast, our developed structures 1-12 (Fig 2) belong to the same class of aminoglycosides as gentamicin, but have no antibacterial activity, exhibit significantly higher readthrough activity and lower toxicity than gentamicin and they consistently demonstrated higher efficiency than that of gentamicin in a series
6
Timor Baasov
of diseases models both in vitro and in vivo tests: compound 2 in cellular and animal models of CF (ref. no. 7 in LP), and cellular models of Rett syndrome (refs. 5, 8); compounds 1 and 2 in cellular and in vivo models of USH1 (refs. 4, 11); compound 4 in cellular and animal models of HS (ref. 10). These observations, together with the relatively low toxicity and high degree of potency of the new generation structures 9-12 in targeting all six different nonsense constructs underlying USH1, CF, DMD and HS, support the feasibility of testing these novel AGs in treating these diseases in animal and human subjects.
3. Comprehensive Description of the Proposed Research
The data presented in the previous sections suggest that the newly developed structures 1-12 (Fig. 2) also named NB-compounds (NBs) are worth for further tests and development as drugs for treatment of cystic fibrosis and other genetic diseases caused by nonsense mutations.
The main objectives of this research proposal are:
(1) to synthesize a large quantities of the selected NB-compounds, namely compound 3 (NB74), compound 4 (NB84) and compound 9 (NB124), and supply them to Eloxx company for the initial Eloxx evaluation to validate our published data on this particular compounds;
(2) to further development NB-compounds for improved cellular uptake, increased bioavailability and acute delivery.
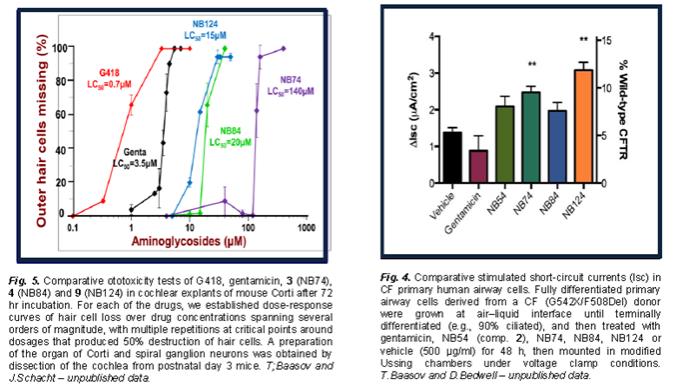
Why we selected especially XX00, XX00 and NB124 for drug development? First, previous studies have shown that the efficiency of aminoglycosides-induced readthrough is highly dependent on: (i) the identity of stop codon (UGA > UAG > UAA), (ii) the identity of the first nucleotide immediately downstream from the stop codon (C > U > A ³ G) and (iii) the local sequence context around the stop codon. Second, while in general the efficiency rank follows NB124 > NB84 > NB74, it is not exactly true for all the mutations and the efficacy might be varied significantly between different mutations for different compounds. For example, our data on CF model shown in Fig. 4 (T. Baasov, X. Xxxx and X. Xxxxxxx, unpublished data) clearly demonstrates that NB74 is more efficient than NB84 to rescue the functional CFTR protein in primary bronchial epithelial cells of a CF donor. Even though NB124 demonstrated highest activity, it is still somewhat more toxic than NB84 and NB74 (Fig. 5), as demonstrated by comparative testing of the potential ototoxoicity of these compounds on the mice cochlear explants (T. Baasov and Xxxxxx Xxxxxxx, unpublished data). It is clear that for the patients with genetic
7
Timor Baasov
diseases requiring a life-long treatment with these compounds, the potential toxicity of these compounds is the most critical factor that limits their further development as potential drug. Therefore, even though in general NB74 is less active than NB84 and NB124, because its low toxicity is definitely worth for further consideration for drug development.
Note that, because its low in vitro activity, NB74 has not pursued for long-term in vivo animal studies. NB84 and NB124 have been evaluated in vivo in various diseases models and have shown to be significantly better than gentamicin.
The proposed working plan includes:
Aim #1: According to our agreement my group will provide Eloxx the important lead compounds XX00, XX00 and NB124, each compound in the quantities of up to 200 mg, chromatographically pure materials in the estimated time frame outlined below. The stocks will undergo the desired biological tests including, readthrough activity assay in dual luciferase reporter (standard USH-R3X mutation) transfected in HEK cells and cytotixicity tests (HEK cells) to validate the published data on these compounds on these specific assays. Plasmids for readthrough activity assay and detailed synthesis and various biological assay protocols will be supplied as well.
Aim #2: My group will continue further development of the lead compounds (NB74, NB84 and NB124) to establish the formulation for increased bioavailability and acute delivery (instead of systemic delivery, e.g. intravenous and/or subcutaneous injection as they have been tested until to date). Shortly, because aminoglycosides (AGs), like gentamicin and our leads XX00, XX00 and NB84 are highly charged, water soluble compounds, they poorly absorb through intestinal tissues and therefore are usually administered by injection. In addition, AGs are short-lived molecules in the circulatory system, being rapidly eliminated by glomerular filtration in the kidney. They also exhibit poor permeability into eukaryotic cells, which requires their administration in higher dosages that in turn causes harmful side effects and limits their use in translational therapy. To solve these problems, we have initiated two different but complementary directions.
Aim #2a: Chemical modification of NBs for increased lipophilicity and better cellular uptake. To address this issue, we already initiated a project in which we examined attachment of various alkyl/aryl groups on the pseudo-disaccharide scaffold of lead structures (e.g. NB74 and NB124) at the N1 position and generated four new scaffolds, compounds 13-16 (Fig. 6, T. Baasov, unpublished data):


Interestingly, the readthrough activity of the previous pseudo-disaccharide scaffolds, compounds 17 (NB82) and 18 (NB83) that provide the basic disaccharide parts of XX00, XX000 (xxx compound 17) and of NB84 (the compound 18) have been significantly improved. Especially noteworthy are the compounds 14 and 16, exhibiting the propyl and benzyl group substitutions at N-1 position, which exhibited highest activity. Compounds 14 and 16 were further tested for their comparative activity against 17 and 18 in various diseases relevant reporter systems (6 reporters that underline the diseases CF, DMD, USH and Hurler syndrome), along with their inhibition of protein synthesis and toxicity (data not shown) and found that these two scaffolds are indeed more active and less toxic than the previously reported
8
Timor Baasov
compounds 17 (NB82) and 18 (NB83). Based on these excellent preliminary data, we aim to use these two newly developed scaffolds, compounds 14 (NB146) and 16 (NB145) to construct new generation of pseudo-trisaccharides similar to our previous leads. These will be done by attachment of riboseamine and 5”-methyl ribosamine at C5 position of the scaffolds to generate four new structures, compounds 19-22 (Fig. 7). The synthesis of the new structures will be performed by using a general tools and methodologies as described in our previous reports. Once the new structures will be available they will be subjected to a series of readthrough, translation inhibition and toxicity tests as these methodologies are already well established in our laboratory.
Aim #2b: Development of new formulation of the lead structures XX00, XX00 and NB124 for increased bioavailability and acute delivery. To address this issue we have initiated a project in which we examined the formulation in which AGs are microencapsulated into nano-cochleates through charge- charge interaction between the AGs and lipid (phosphatidilserine-PS). We also developed an efficient ELISA assay method to detect the encapsulated AG drug in cochleates and/or in biological fluids (serum, urine, tissue, etc.). The preliminary data on comparative readthrough activity of the clinical drugs gentamicin and G418 are attached below as Appendix 1. As can be seen from these data, gentamicin-cochleate preparation is far better in terms of readthrough activity than the corresponding gentamicn drug in solution. Further steps include: a) to compare and contrast XX00, XX00 and NB124 in drug-cochleate versus solution in terms of readthrough efficiency in dual luciferase assay protocol; b) the best formulations from the previous step will be further evaluated for toxicity and rescue of functional CFTR protein (in collaboration with the University of Alabama at Birmingham) in cell lines and in CF-mouse models. c) To determine the structure/size of the resulted drug-cochleate complexes by various spectroscopic methods including cryo-electron-microscopy and freazfraction-microscopy techniques. d) Continueous structure-activity relationship study to develop appropriate size nano-cochleates with maximum activity and lowered toxicity suitable for acute administration to treat genetic diseases (this part of the project will be done in collaboration with Xxxx. Xxxxxx Xxxxxx of the Biotechnology and Food Engineering Department of Technion).

Note that the labs of Profs. Xxxxxxx and Xxxxxxx are already tightly collaborating with my lab and we have series of joint publications on this subject (Xxxxxxx: refs 7, 10 and Xxxxxxx: J.Med.Chem. 2009, 52, 2836- 2845).
4. Proposed Budget (in US Dollars) and Time Frame
| Compounds |
Time Frame |
Budget Requested |
Remarks |
Notes | ||||||||
| Aim # 1 | PI T. Baasov | |||||||||||
| ~200 mg NB74 | 2 weks-1 month | $ | 8,000 | Rapid initial supply | ||||||||
| ~200 mg NB84 | 2-3 months | $ | 10,000 | Rapid initial supply | ||||||||
| ~200 mg NB124 | 2-3 months | $ | 12,000 | Rapid initial supply | ||||||||
| Total Aim #1: | $ | 30,000 | ||||||||||
|
|
|
|
||||||||||
| Aim # 2a | ||||||||||||
| Synthesis of 19-22 and their evaluations | 8-12 months | $ | 40,000 | |||||||||
| Aim # 2b | ||||||||||||
| Preliminary studies for standard drug-cochleates SAR | 4-6 months | $ | 10,000 | The cost of PS and the synthesis of NBs are included | ||||||||
| NB84-cochleates SAR | 4-6 months | $ | 15,000 | |||||||||
| Total Aim #2: | $ | 65,000 | ||||||||||
|
|
|
|
||||||||||
| Grand total for the Aims # 1&2: |
$ | 95,000 | ||||||||||
|
|
|
|
||||||||||
9
Timor Baasov
5. Detailed Budget (in US Dollars)
Personnel:
| Role in project |
% |
Time | Salary | |||||||||
| 1. |
Lab. Assistant | Lab. Assistant | 50 | 12,000 | ||||||||
| 2. |
Postdoctorant | Researcher | 50 | 12,000 | ||||||||
|
|
|
|||||||||||
| Total: | 24,000 | |||||||||||
|
|
|
|||||||||||
Supplies:
| 1. Chemicals, absolute & deuterated solvents |
28,000 | |||
| 2. Lab. equipment, glassware, plastic xxxx |
12,000 | |||
| 3. Biochemicals |
2,000 | |||
| 4. In vitro and ex vivo assay kits |
10,000 | |||
| 5. Chromatography material for various purifications |
13,000 | |||
| 6. NMR and Mass Spectrometry tests |
6,000 | |||
| Total: |
71,000 | |||
|
|
|
|||
| Grand Total: |
$ | 95,000 | ||
|
|
|
Budget Justification:
The requests for laboratory assistant and postdoctorant for the duration of the grant period are in recognition of the amount of work required in this project. Considerable effort will be expended in the syntheses of various lead compounds discussed in the proposal, their structure determination and analysis, assays for their activity. The request for materials, supplies, and chemicals/biochemicals is an important part for a successful development and completion of the project.
6. Investigators’ Curriculum Vitae
Surname: Baasov First name: Timor
Birthdate: January 3, 1954
(a) Education Background
| From-To |
Institution |
Area of specialization |
Degree | |||
| 1981-1986 |
Weizmann Institute of Science | Chemistry | Ph. D. | |||
| 1977-1979 |
Tel-Aviv University | Chemistry | M. Sc. | |||
| 1975-1977 |
Tel-Aviv University | Chemistry | B. Sc. |
Major research interest: Carbohydrate chemistry, Bioorganic and medicinal chemistry, Drug design and development, Rational design of substrate and inhibitors, Mechanistic enzymology.
10
Timor Baasov
(b) Employment
| From-To |
Institution |
Research area |
Title | |||
| 2004- |
Technion | Bioorganic Chemistry | Professor | |||
| 3/1998-8/1998 |
The Scripps Research Institute, Cal | Bioorganic Chemistry | Visiting Prof. | |||
| 1998-2003 |
Technion | Bioorganic Chemistry | Assoc. Prof. | |||
| 1990-1998 |
Technion | Bioorganic Chemistry | Senior Lecturer | |||
| 1988-1990 |
Technion | Bioorganic Chemistry | Lecturer | |||
| 1986-1988 |
Harvard University | Bioorganic Chemistry | Post-Doct. Res. |
11
Timor Baasov
7. List of Publications (2010-2112)
| 1. | X. Xxxxxxxx, X. Xxxxxx, X. Xxxxxxx, X. Xxxxxxxxxxx, X. Xxxxxxxx and X. Xxxxxx. Repairing faulty genes by amino glycosides: Development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg. Med. Chem. 18, 3735-3746, (2010). |
| 2. | V. Pokrovskaya, X. Xxxxxxxx, X. Xxxxxxxxx and X. Xxxxxx. Aminoglycosides: Redesign Strategies for Improved Antibiotics and Compounds for Treatment of Human Genetic Diseases. Methods in Enzymology. 478 (Glycomics), 437-462, (2010). |
| 3. | V. Pokrovskaya and T. Baasov. Dual-acting hybrid antibiotics: a promising strategy to combat bacterial resistance. Expert Opinion in Drug Discovery. 5(9), 883-903, (2010). |
| 4. | X. Xxxxxxx, X. Xxxxxx-Xxxxxx, X. Xxxxxxxx, X. Xxxxxxxx, X. Xxxxxxxx, X. Xxxxxx, T. Xxx-Xxxxx, X. Xxxxxxx and X. Xxxxx-Xxxxxxx. Designed aminoglycoside NB30 induces beneficial read-through of a USH1C nonsense mutation in the retina. Investigative Ophthalmology & Visual Science, 51(12), 6671-6680, (2010). |
| 5. | X. Xxxxxxx, X. Xxxxxxxx, X. Xxxxxx, X. Xxxxxxx, X. Xxxxxxxx, X. Xxxxxxxx, X. Xxxxxx, X. Xxxxxx. Readthrough of Nonsense Mutations in Rett Syndrome: Evaluation of novel aminoglycosides and generation of a new mouse model. Journal Molecular Medicine, 89, 389-398, (2011). |
| 6. | X. Xxxxxxxxx, X. Xxxx-Xxxxxx, X. Xxxxxxxx, X. Xxxxxx. Repairing faulty genes by aminoglycosides: Identification of new pharmacophore with enhanced suppression of diseases-causing nonsense mutations. Medicinal Chemistry Communications, 2, 165-171 (2011). |
| 7. | X.X. Xxxx, X.X. Xxxx, X. Xxxxxx, X. Xxxxxx, X. Xxxxx, X. Xxxx;xx-Xxxxxxx, X. Xxxxxxxx, X. Xxxxxxxx, X. Xxxxx, X. Xxxxxxxxxx, X. Xxxxxx, X.X. Xxxxxxx. Suppression of CFTR Premature Termination Codons and Rescue of CFTR Protein and Function by the Synthetic Xxxxxxxxxxxxxx XX00. Journal Molecular Medicine (Berl), 89, 1149-1154 (2011). |
| 8. | X. Xxxxxxx, B. Xxx Xxxx, X. Xxxxxxxx, X. Xxxxxxxx, X. X. Xxxxx, X. Xxxxxxxxx, G. Rechavi, T. Baasov, E. Gak. Ex Vivo Treatment with a Novel Synthetic Aminoglycoside NB54 in Primary Fibroblasts from Rett Syndrome Patients Suppresses MECP2 Nonsense Mutations. PLoS ONE, 6 (6), e20733 (2011). |
| 9. | H-L. X. Xxx, C-C. Xxxx, X. Xxxxxx, Y. Xxx, X. X. Xxxxxxxxx. Post-transcriptionally Regulated Expression System in Human Xenogeneic Transplantation Models. Molecular Therapy, 19(9), 1645-1655 (2011). |
| 10. | X. Xxxx , X. Xxxxxxxx, X. Xxxxxxxxx, X. Xxxxxx, S-C. Li, Y-T Li, X.X. Xxxxxxx, X.X. Xxxxxxx. The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse. Molecular Genetics and Metabolism 105, 116-125 (2012). |
| 11. | X. Xxxxxxxx, X. Xxxxxxxx, X. Xxxxxx, X. Xxxxxxxx, M. van Wyk, T. Baasov, X. Xxxxxxx, and X. Xxxxx-Xxxxxxx. A comparative evaluation of XX00, XX00 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Molecular Medicine, 4, 1-14, (2012). |
| 12. | X. Xxxxxxxxx, X. Xxxx-Xxxxxx, X. Xxxxxxx, X. Xxxxxxx, X. Xxxxxx, X. Xxxxxxxx X. Xxxxxx. Increased Selectivity toward Cytoplasmic versus Mitochondrial Ribosome Confers Improved Efficiency of Synthetic Aminoglycosides in Fixing Damaged Genes: A Strategy for Treatment of Genetic Diseases Caused by Nonsense Mutations. J. Med. Chem. 55(23), 10630-10643 (2012). |
| 13. | M. Schalev, X. Xxxxxxxxx, X. Xxxxxx, X. Xxxxxxxx, X. Xxxxx-Xxxxxxxxx, X. Xxxxxx. Development of generic immunoassay for the detection of a series of aminoglycosides with 6’-OH group for the treatment of genetic diseases in biological samples. Journal of pharmaceutical and biomedical analysis. 75, 33-40 (2013). |
| 14. | X.X. Xxxxxxx, X. Xxxx, X. Xxx, X. Xxxxxxxxx, X. Xxxxxx, X. Xxxxx; X. Xxxxxxxx, X. Xxxxxxxxx, X.X. Xxxx, X. Xxxxxx, X.X. Xxxxxxx. Attenuation of Nonsense-Mediated mRNA Decay Enhances In Vivo Nonsense Suppression. PLoS ONE 8 (4), e60478 (2013). |
| 15. | M. Schalev, X. Xxxxx, D. Kopelyanskiy, X.X. Xxxxx, X. Xxxx, X. Xxxxxx. Identification of the molecular attributes required for Aminoglycoside activity against Leishmania. PNAS 110 (33), 13333-13338 (2013). |
| 16. | X. Xue, X. Xxxxxx, X.X. Xxxx, X. Xxxxxx, M. Du, L. A. Jackson, Y. Dai, X. Xxxxxxxx, X. Xxxxxx, X. Xxxx, X. Xxxxxxx, X. Xxxxxxx, X. Xxxxxx, X. Xxxx, X. X. Xxxxxxx, X.X. Xxxx. Synthetic Aminoglycosides Efficiently Suppress CFTR Nonsense Mutations and Are Enhanced by Ivacaftor. Submitted (2013). |
| 17. |
12
Appendix I
Comparative ex-vivo read through activity of aminoglycosides between the solution and encapsulated within phosphatydylserine multilamelar structures (cochleates)

Empty-C: Phosphatidyserine (PS) based cochleates has been prepared in the absence of an aminoglycosidic agent (CaCl2 was used to replace the cation required for cochleation procedure). Genta/G418: in solution. Genta-C/G-418-C: Gentamicin or G418 were encapsulated within PS based cochleates.
Ex-vivo suppression of the R3X (USH1) mutation. The constructs of p2luc plasmid harboring the R3X mutation were transfected to HEK-293 cells and addition of the tested compounds was performed 6 h post transfection. Luciferase activity was determined using the Dual Luciferase Reporter Assay System (Promega™).
Each bar represents the mean ± S.E.M. of 3 independent experiments (2 duplicated each). Bars that are marked in an asterix (*) sign differ significantly at p < 0.05 according to a paired t test analysis.
Timor Baasov
Appendix II
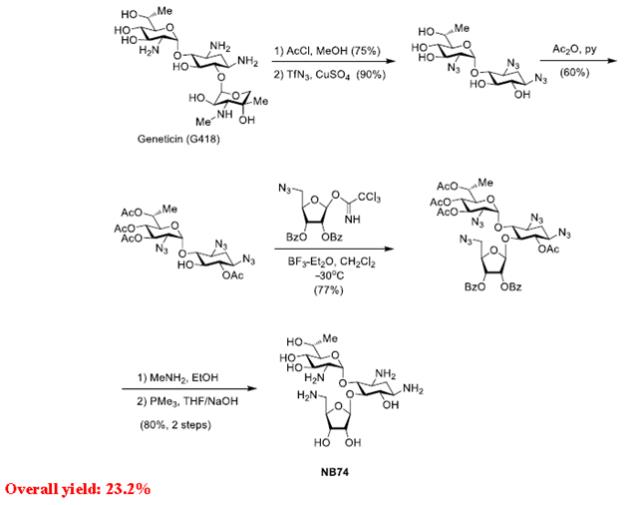
Synthetic schemes for the assembly of the developed leads NB74 and NB84 along with of the required donor structure
Synthesis of NB74
14
Timor Baasov
Synthesis of NB84

15
Timor Baasov
Preparation of Trichloroacet-imidate DONOR (for both NB74 and NB84)

Note: All the synthetic procedures and analytical data for the above syntheses of XX00, XX00 and NB124 are published in the Bioorganic and Medicinal Chemistry Paper (2010) and JMC (2012).
16

