DEVELOPMENT, LICENSE AND COMMERCIALIZATION AGREEMENT
Exhibit 10.4
Confidential Materials omitted
and filed separately with the
Securities and Exchange Commission. Asterisks denote omissions.
CONFIDENTIAL
DEVELOPMENT, LICENSE AND COMMERCIALIZATION AGREEMENT
This DEVELOPMENT, LICENSE AND COMMERCIALIZATION AGREEMENT (“Agreement”) is entered into between Sepracor Inc., a company organized under the laws of the State of Delaware, United States, and having its principal place of business at 00 Xxxxxxxxx Xxxxx, Xxxxxxxxxxx, XX 00000-0000, Xxxxxx Xxxxxx, and Glaxo Group Limited, a company organized under the laws of England & Wales and having its principal place of business at Glaxo Xxxxxxxx Xxxxx, Xxxxxxxx Xxxxxx, Xxxxxxxxx, Xxxxxxxxx, XX0 0XX, Xxxxxx Xxxxxxx (“GSK”).
A. Sepracor has conducted non-clinical and clinical trials, and has been granted Marketing Approval by the FDA to commercialize, and is commercializing in the United States, the LUNESTA® eszopiclone-based insomnia product; and
B. Sepracor owns or controls certain patents, know-how, trademarks and other intellectual property relating to the Product in the GSK Territory; and
C. GSK is experienced in developing, registering, securing pricing approvals, marketing and distributing pharmaceutical products in the GSK Territory; and
D. On the terms and conditions set forth in this Agreement:
1. Sepracor and GSK wish to collaborate on the potential for further development (if any), as well as the registration and commercialization of the Product in the GSK Territory;
2. Sepracor is willing to grant to GSK, and GSK desires to obtain from Sepracor, certain exclusive rights and licenses with respect to the potential development (if any), registration and commercialization of the Product in the GSK Territory; and
3. Sepracor is willing to supply GSK with Bulk Tablets of the Product for packaging and commercial sale by GSK in the GSK Territory.
NOW, THEREFORE, in consideration of the foregoing premises and the mutual covenants herein contained, and for other good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, Sepracor and GSK hereby agree as follows:
CONFIDENTIAL
ARTICLE I.
DEFINITIONS
As used in this Agreement, the following terms shall have the meanings set out in this Article 1 unless the context clearly and unambiguously dictates otherwise.
1.1. “Active Ingredient” shall mean the chemical compound known as eszopiclone, also identified by the chemical name (+) 6-(5-chloro-2-pyridinyl)-6,7-dihydro-7-oxo-5H-pyrrolo [3,4b] pyrazin- 5-yl 4-methylpiperazine-1-carboxylate or (+) 6-(5-chloropyri-2-dyl)-5-(4-methyl piperazin-1-yl) carbonyloxy-7-oxo-6,7-dihydro-5H-pyrrolo [3,4b] pyrazine, and all pharmaceutically active derivatives thereof, such as, but not limited to, its metabolites, and all esters, salts, hydrates, solvates, polymorphs and isomers of all the above.
1.2. “Actual Cost” shall mean, the fully loaded cost incurred by Sepracor to manufacture or have manufactured Bulk Tablets, including for avoidance of doubt the overhead associated with performing its obligations, as may be agreed by the parties, under the Supply Agreement. The Actual Cost may be more specifically defined in the Supply Agreement, consistent with Schedule 10.2 herein, and shall be documented by Sepracor to GSK to the reasonable satisfaction of GSK.
1.3. “Affiliate” of a Party shall mean any person, corporation or other entity that, directly or indirectly, through one or more intermediaries, controls, is controlled by, comes to control, or is under common control with such Party, as the case may be. As used in this Section 1.3, “control” shall mean: (i) to possess, directly or indirectly, the power to direct the management and policies of such person, corporation or other entity, whether through ownership of voting securities or by contract relating to voting rights or corporate governance; or (ii) direct or indirect beneficial ownership of at least fifty percent (50%) (or such lesser percentage which is the maximum allowed to be owned by a foreign corporation in a particular jurisdiction) of the voting share capital in such person, corporation or other entity.
1.4. “Acquired Competing Product” shall mean any product, in respect of which GSK or its Affiliates, during the Term, acquires the rights (by license, purchase or otherwise) to promote, market or sell in the GSK Territory, which at the time of acquisition: (i) [**]; (ii) [**]; or (iii) [**].
1.5. “Acquisition Transaction” shall mean the acquisition of the assets of a Third Party, whether through merger, purchase of stock representing fifty percent (50%) or more of the outstanding voting stock, purchase of all or substantially all of the assets of such entity or otherwise.
1.6. “Blocking Patent” means any Patent issued to a Third Party that would be infringed by the manufacture, use, sale, offer for sale or importation of the Product in the GSK Territory.
CONFIDENTIAL
1.7. “Bulk Tablets” shall mean finished tablets containing the Active Ingredient which meets the relevant specifications, to be defined more specifically in the Supply Agreement, and to be shipped to GSK by Sepracor in bulk in the manner set forth in the Supply Agreement, and then to be packaged as the Product by or on behalf of GSK.
1.8. “Business Day” shall mean a day other than a (i) Saturday, (ii) Sunday or (iii) public holiday in the United States or England. For the avoidance of doubt, references in this Agreement to “days” shall mean calendar days.
1.9. “Calendar Quarter” shall mean a period of three (3) consecutive months during a Calendar Year beginning on and including 1st January, 1st April, 1st July or 1st October.
1.10. “Calendar Year” shall mean a period of twelve (12) consecutive months beginning on and including 1st January.
1.11. “Clinical Supplies” shall mean those quantities of Bulk Tablets and placebo to be supplied by Sepracor to GSK in the manner to be set forth in the Supply Agreement for use in the following scenarios where the Bulk Tablets are not to be packaged and labeled for commercial sale: (i) any Developmental Study; (ii) validation studies or any other studies to confirm packaging or formulation or other manufacturing requirements; or (iii) for any other non-commercial uses necessary for obtaining the Marketing Approval for the Product in any country in the GSK Territory.
1.12. “Commercialization Studies” shall mean any clinical studies GSK wishes to be conducted by or on behalf of GSK in the GSK Territory in support of GSK’s commercialization of the Product in the GSK Territory and which will be initiated in a country after Marketing Approval of the Product in such country. Such studies may include, but shall not be limited to, “marketing studies” to support market uptake, acceptance and differentiation of the Product, epidemiological studies, modeling and pharmacoeconomic studies, and studies in support of modifications or additional sleep disorder related indications. Commercialization Studies shall not include studies designed to generate data in support of new indications unrelated to sleep disorders, or studies investigating the safety or efficacy of the Product in a modified release formulation or a combination product. For the avoidance of doubt, Commercialization Studies shall not include Required Studies, Investigator Sponsored Clinical Studies, Sepracor Studies, studies Sepracor initiated prior to the Effective Date in support of the Product, any studies relating to the Product that Sepracor may initiate in the Sepracor Territory or any other studies that Sepracor may initiate anywhere in the world which do not relate to the Product.
1.13. “Commercially Reasonable Efforts” means those diligent efforts and resources, with respect to a particular Party, at the relevant point in time, that are comparable to those generally used by that Party, in good faith, in the exercise of its reasonable and prudent scientific and business judgment relating to other prescription pharmaceutical products owned or licensed by it or to which it has exclusive rights, which have market potential and are at a stage of development or product life similar to the Product, taking into account measures of relative safety and efficacy, product profile, the competitiveness of the marketplace, the proprietary
CONFIDENTIAL
position of the compound or product, the regulatory structure involved, the relative profitability of the products, and other relevant factors, including without limitation comparative technical, legal, scientific, and/or medical factors. Such Commercially Reasonable Efforts shall include, without limitation, comparable efforts with respect to (i) promptly assigning responsibility for development and commercialization activities to specific employees who are held accountable for progress and monitoring such progress on an on-going basis, (ii) setting and consistently seeking to achieve specific and meaningful objectives and timelines for carrying out such development and commercialization activities, (iii) consistently making and implementing decisions and allocating resources designed to advance progress with respect to such objectives and timelines, and (iv) employing compensation systems for its employees working with the Product that are similar to the compensation systems the applicable Party applies with respect to its other programs with products of similar potential.
1.14. “Confidentiality Agreement” shall mean that certain letter agreement dated January 11, 2005 entered into by and between Sepracor and GlaxoSmithKline Research & Development Limited.
1.15. “Control” (including any variations such as “Controlled” and “Controlling”), in the context of intellectual property rights and Know-How, shall mean rights to intellectual property sufficient to grant the applicable license under this Agreement, without violating the terms of an agreement with a Third Party.
1.16. “Core Campaign Product Strategies” shall mean those materials proposed by the GSK European marketing group which outline the core marketing materials to be used in relation to the Product and to specifically include trade dress, core claims and visuals.
1.17. “Data” shall mean, subject to the provisions of Section 5.10, any and all research data, pharmacology data, preclinical data, clinical data, manufacturing data and/or all regulatory documentation, information and submissions pertaining to, or made in association with, an IND or a Marketing Authorization Application for the Product, in each case which are Controlled by a Party as of the Effective Date or during the Term of this Agreement, including but not limited to the Development Data.
1.18. “Developmental Studies” means any (i) Required Studies or (ii) Commercialization Studies, both as set forth in the Development Plan. For the avoidance of doubt, Developmental Studies shall not include Investigator Sponsored Clinical Studies of the Product in the GSK Territory.
1.19. “Effective Date” shall mean the later of: (i) the date on which an authorized representative of Sepracor executes this Agreement, and (ii) the date on which an authorized representative of GSK executes this Agreement.
1.20. “EMEA” shall mean the European Medicines Agency, or any other successor agency that is responsible for approving the sale of pharmaceutical products across the European Union.
CONFIDENTIAL
1.21. “European Union” shall mean the member countries of the European Union at the relevant point in time.
1.22. “FDA” shall mean the United States Food and Drug Administration, or any successor entity thereto performing similar functions.
1.23. “GSK Competing Product” shall have the meaning stated in Section 9.1(e) of this Agreement. For the avoidance of doubt, GSK Competing Products may include, but shall not be limited to, compounds, other than Acquired Competing Products, acquired by GSK (by licence, purchase or otherwise) during the Term of this Agreement.
1.24. “GSK Territory” shall mean all countries and territories of the world excluding the Sepracor Territory.
1.25. “Improvements” shall mean any new or useful process, technique, formula, invention or know-how, formulation, or composition of matter relating to or comprising the Product, or any Sepracor Technology, whether patentable or unpatentable, or any improvement, enhancement, modification or derivative work thereof, that is conceived or first reduced to practice or first demonstrated to have utility during the Term of this Agreement in connection with the Parties’ activities under this Agreement.
1.26. “IND” shall mean an Investigational New Drug Application (including any amendments thereto) filed with the FDA pursuant to 21 C.F.R. §312 before commencement of clinical trials of a pharmaceutical product, or any comparable filings with any Regulatory Authority in any other jurisdiction, including, but not limited to, clinical trial applications.
1.27. “International Financial Reporting Standards” shall mean the International Financial Reporting Standards and International Accounting Standards issued by the International Accounting Standards Board (“IASB”) and interpretations issued by the International Financial Reporting Interpretations Committee of the IASB.
1.28. “Major Market” shall mean each of the U.K., Germany, Italy, France, Spain, China, Australia, Brazil and South Korea, provided, however, with respect to Sections 6.1, 6.3(l), 6.4(b), 14.4(c) and 16.2(e), Major Markets shall only include U.K., Germany, Italy, France and Spain. A country shall no longer be included in the definition of Major Market for the purposes of Sections 2.1(b), 6.1, 6.3(l), 7.3(d) and 16.2(e) once a Generic Product has been launched in such country.
1.29. “Marketing Approval” shall mean all approvals, licenses, registrations or authorizations of Regulatory Authorities in a country necessary for the manufacture, use, storage, import, export, distribution, promotion, marketing, offer for sale and sale of the Product in such country. For countries where governmental approval is required for pricing and/or reimbursement for the Product to be reimbursed by national health insurance (or its local equivalent), “Marketing Approval” shall not be deemed to occur until such pricing and/or reimbursement approval is obtained.
CONFIDENTIAL
1.30. “Marketing Authorization Application” (or “MAA”) shall mean a filing, comparable to a New Drug Application (as defined in 21 C.F.R. § 314.50 et. seq.), for Marketing Approval (not including pricing or reimbursement approval) for the Product in a country within the GSK Territory.
1.31. “Net Sales” shall mean the gross amounts received for sales of the Product by or on behalf of GSK, its Affiliates and/or Sublicensees (the “Selling Party”) to Third Parties, less deductions actually allowed or specifically allocated to the Product by the Selling Party using International Financial Reporting Standards for:
(a) transportation charges to the extent that they are included in the price or otherwise paid by the purchaser, including, without limitation, insurance, for transporting Product and separately identified on the invoice or in other documentation maintained in the ordinary course of business; and;
(b) trade, quantity and cash discounts, or charge-backs, refunds or other rebates actually granted to the customer (including, if applicable, hospitals or private or public health insurance entities);
(c) credits, rebates and allowances to the customer on account of rejection or returns of the Product (including wholesaler and retailer returns), or on account of non-discretionary retroactive price reductions affecting such Product;
(d) sales and excise taxes, other consumption taxes, customs duties and customary compulsory payments to governmental authorities and any other governmental charges imposed upon the production, importation, use or sale of the Product actually paid and separately identified on the invoice or in other documentation maintained in the ordinary course of business; and
(e) any other items actually deducted from gross invoices sales amounts as reported by the Selling Party in its financial statements in accordance with the International Financial Reporting Standards, applied on a consistent basis
In no event will any particular amount, identified above, be deducted more than once in calculating Net Sales (i.e., no “double counting” of reductions). Sales of the Product between GSK and its Affiliates or Sublicensees shall be excluded from the computation of Net Sales, but the subsequent resale of such the Product to a Third Party shall be included within the computation of Net Sales.
In the case of any sale or disposal for value, other than in an arms length transaction exclusively for money, such as barter or counter trade, Net Sales shall be calculated as above on the value of the consideration received or the fair market value (if higher) of the Product in the country of sale or disposal.
1.32. “Non-Severable Improvements” shall mean an Improvement to the Sepracor Technology which is incapable of exploitation independently of the Sepracor Technology
CONFIDENTIAL
Controlled by Sepracor or its Affiliates as of the Effective Date and during the Term of this Agreement.
1.33. “Party” shall mean Sepracor or GSK individually, and “Parties” shall mean Sepracor and GSK collectively.
1.34. “Patent(s)” shall mean patents and patent applications, together with all additions, divisions, continuations, continuations-in-part, provisionals, substitutions, reissues, re-examinations, extensions, registrations, patent term extensions, supplemental protection certificates and renewals of a patent or patent application, and any confirmation patent or registration patent or patent of addition based on any such patent.
1.35. “Person” shall mean any individual, corporation, partnership, limited liability company, trust, governmental entity, or other legal entity of any nature whatsoever.
1.36. “Phase III Clinical Trial” shall mean a human clinical trial, the principal purpose of which is to gather safety and efficacy data for a Product Indication of one or more particular doses in patients being studied that is needed to evaluate the overall benefit and risk relationship of the product and to provide adequate basis for labeling, as required in 21 C.F.R. §312(c), or such similar clinical study in a country other than the United States of America.
1.37. “Product” shall mean the Active Ingredient, in the form and dosages approved as of the Effective Date by the FDA as LUNESTA® (eszopiclone), for all indications which are or may be approved by Regulatory Authorities in the GSK Territory for those forms and dosages. The Product shall also include any additional immediate release forms or dosages of LUNESTA® (eszopiclone) that may be developed by Sepracor or GSK during the term of the Agreement.
For the avoidance of doubt, Product shall not mean, inter alia: (i) any product containing Active Ingredient in [**]; or (ii) any product with the Active Ingredient used [**]. All such products to be referred to herein as “Excluded Products”.
1.38. “Product Indication” shall mean, on a country-by-country basis, any indication for which the Product has Marketing Approval in such country.
1.39. “Product Trademarks” shall mean the trademark(s) owned by Sepracor which are available for use with the Product, as listed in Part A of Schedule 1.39.
1.40. “Regulatory Authority” shall mean the EMEA and any other national, regional, state or local regulatory agency, department, bureau, commission, council or other governmental entity whose review and/or approval is necessary for the manufacture, packaging, use, storage, import, export, distribution, promotion, marketing, offer for sale and sale of the Product in the GSK Territory. For countries where governmental approval is required for pricing or reimbursement for the Product to be reimbursed by national health insurance (or its local equivalent), “Regulatory Authority” shall also include any national, regional, state or local
CONFIDENTIAL
regulatory agency, department, bureau, commission, council or other governmental entity whose review and/or approval of pricing or reimbursement is required.
1.41. “Required Studies” shall mean any pre-clinical or clinical studies (other than those initiated by Sepracor prior to the Effective Date) that are required or requested in writing by any Regulatory Authority as a condition of, or in connection with, obtaining Marketing Approval for the Product in one of more countries in the GSK Territory.
1.42. “Sepracor” shall mean Sepracor Inc. and its Affiliates.
1.43. “Sepracor Know-How” shall mean, subject to Section 5.10 and the last sentence of Section 7.6, all present and future scientific, medical, technical, manufacturing, regulatory and other information relating to the Product (including results of any Developmental Study and Data), which are owned or Controlled by Sepracor as of the Effective Date or during the Term of this Agreement. Sepracor Know-How shall include all such items that are generated by or under authority of Sepracor during the Term of this Agreement.
1.44. “Sepracor Patents” shall mean, subject to Section 7.6, all present and future Patents owned or Controlled by Sepracor, which generically or specifically claim Product, a composition comprising Product, or a use of Product. The current list of Patents is set forth in Schedule 1.44, and such list shall be updated by Sepracor during the Term of the Agreement, at least on a semi-annual basis.
1.45. “Sepracor Studies” shall mean any pre-clinical or clinical studies (other than those initiated by Sepracor prior to the Effective Date) that Sepracor wishes to conduct or have conducted for it in the GSK Territory related to: (i) the Product; or (ii) the Excluded Products. For the avoidance of doubt, this does not include any study Sepracor may conduct or have conducted for it outside of the GSK Territory.
1.46. “Sepracor Technology” shall mean, subject to Section 7.6, all Sepracor Know-How, and Sepracor Patents, which are Controlled by Sepracor or its Affiliates, prior to the Effective Date, and all Improvements Controlled by Sepracor or its Affiliates during the Term of this Agreement, if any. Sepracor Technology excludes Product Trademarks and Other Trademarks.
1.47. “Sepracor Territory” shall mean Canada, United States (and its incorporated and unincorporated territories), Mexico and Japan.
1.48. “Severable Improvements” shall mean an Improvement to the Sepracor Technology which is capable of exploitation independently of the Sepracor Technology Controlled by Sepracor or its Affiliates as of the Effective Date and during the Term of this Agreement.
1.49. “Sublicensee” shall mean a Third Party approved by Sepracor, or a GSK Affiliate, to whom GSK, under the terms of this Agreement, has granted a right to sell, market, distribute
CONFIDENTIAL
and/or promote the Product in the GSK Territory and “Sublicense” shall mean an agreement or arrangement between GSK and a Sublicensee granting such rights. As used in this Agreement, Sublicensee shall not include (i) a wholesaler, distributor or reseller of the Product who does not market or promote the Product, (ii) a contract manufacturer or packager or (iii) a contract research organization.
1.50. “Target Label” shall mean approved labeling for the Product with: (i) an indication for the treatment of insomnia; (ii) [**] which would [**], and (iii) [**] attached at Schedule 1.50. For the avoidance of doubt, the label requirements at Section 1.50(ii), above, shall not apply to [**].
1.51. “Term” shall mean the period described in Section 14.1 of this Agreement.
1.52. “Third Party” shall mean any Person other than Sepracor, GSK and their respective Affiliates.
1.53. “Third Party Technology” means any Patents, know-how, inventions, or other intellectual property owned or controlled by a Third Party but not owned or Controlled by a Party or its Affiliates.
1.54. “Transition Period” shall mean the period of time beginning on the Effective Date of the Agreement and ending on the earlier of (i) the date of final approval of a MAA filed with the EMEA; or (ii) a date indicated in a written agreement by Sepracor and GSK.
1.55. “Valid Claim” shall mean a claim of any issued, unexpired patent that has not been revoked or held unenforceable or invalid by a decision of a court or governmental agency of competent jurisdiction from which no appeal can be taken, or with respect to which an appeal is not taken within the time allowed for appeal, and that has not been disclaimed or admitted to be invalid or unenforceable through reissue, disclaimer or otherwise.
1.56. “VAT” shall mean the tax imposed by Council Directive 2006/112/EC of the European Community and any national legislation implementing that directive together with legislation supplemental thereto and in particular, in relation to the United Kingdom, the tax imposed by the Value Added Tax Act of 1994 or other tax of a similar nature imposed elsewhere instead of or in addition to value added tax.
1.57. “Wind-down Period” shall mean any period after the date of termination of this Agreement, in its entirety or on a country-by-country basis, during which pursuant to Sections 15.2(a) or 15.3(a), GSK is required to continue to perform certain activities.
1.58. Other Definitions. Each of the following defined terms shall have the meaning set forth in the indicated Section of this Agreement:
CONFIDENTIAL
|
Defined Term |
|
Section |
|
Acquired Competing Product |
|
9.1(b) |
|
Agreement |
|
Introduction |
|
Commercialization Plan |
|
6.1 |
|
Confidential Information |
|
11.1 |
|
Commercialization Option |
|
9.1(e) |
|
Company RA Executives |
|
7.2(c)(i) |
|
Development Data |
|
11.2 |
|
Development Plan |
|
5.6 |
|
Disclosing Party |
|
11.1 |
|
DMF |
|
5.10(a) |
|
Excluded Products |
|
1.37 |
|
Fault of GSK |
|
17.4(b)(ii) |
|
Fault of Sepracor |
|
17.4(b)(i) |
|
Generic Product |
|
7.3(c)(ii) |
|
GSK Indemnities |
|
17.2 |
|
Indemnitee |
|
17.3 |
|
Indemnitor |
|
17.3 |
|
Independent RA Expert |
|
7.2(c(i) |
|
Infringing Product |
|
12.3 |
|
Infringing Trademark |
|
12.4 |
|
Initial Commercialization Plan |
|
6.1 |
|
Intervening Event |
|
19.1 |
|
Investigator Sponsored Clinical Studies |
|
5.5 |
|
JSC |
|
3.1 |
|
Liabilities |
|
17.1 |
|
Other Trademarks |
|
13.1 |
|
Pharmacovigilance Agreement |
|
5.14(c) |
|
Product Liability Claim |
|
17.5(a) |
|
Quality Agreement |
|
10.2 |
|
Recall Costs |
|
17.4(b) |
|
Receiving Party |
|
11.1 |
|
Reconciliation Report |
|
7.3(e)(ii) |
|
Reconciliation Review Period |
|
7.3(e)(ii) |
|
SEC |
|
11.6 |
|
Selling Party |
|
1.31 |
|
Senior Executives |
|
3.4(b) |
|
Sepracor Development Plan |
|
5.8 |
|
Sepracor Indemnitees |
|
17.1 |
|
Subcommittee |
|
3.2 |
|
Supply Agreement |
|
10.2 |
|
Third Party Claim |
|
17.1 |
|
Trademark Infringing Product |
|
12.4 |
CONFIDENTIAL
ARTICLE II.
GRANT OF LICENSE
2.1. Licenses.
(a) Sepracor hereby grants to GSK an exclusive (even as to Sepracor), royalty-bearing license throughout the GSK Territory under the Sepracor Technology to use, sell, have sold and import the Bulk Tablets and Product. The license shall include, but not be limited to the rights to: (i) purchase Clinical Supplies and the Product in Bulk Tablets for use in the GSK Territory in accordance with this Agreement and the Supply Agreement, (ii) package and/or have packaged the Bulk Tablets, (iii) conduct the activities set forth in the Commercialization Plan with respect to the Product, (iv) offer for sale, sell and distribute the Product, and (v) market and promote the Product under the Product Trademarks and Other Trademarks.
(b) GSK has the right to grant sublicenses under this Agreement to its Affiliates. Except as otherwise provided in Section 19.10, GSK shall not have the right to sublicense, transfer or assign the rights granted under this Agreement to a Third Party without Sepracor’s prior written consent, which shall not be unreasonably withheld or delayed, provided that prior written consent shall not be required in relation to any sublicense of rights territorially limited to those countries in the GSK Territory other than the Major Markets. Where a sublicense of any of the rights or obligations is permitted hereunder, each Sublicensee must enter into a Sublicense Agreement, which must be consistent with the terms and conditions of this Agreement. GSK shall also enforce the performance by any Sublicensee of the terms of each such Sublicense Agreement in order to ensure that any Sublicensee complies with the applicable terms and conditions of this Agreement. The grant of any such sublicense to an Affiliate or a Third Party will not relieve GSK of its obligations under this Agreement, except to the extent they are satisfactorily performed by such Sublicensee and GSK hereby guarantees the satisfactory performance of GSK’s obligations hereunder where performed by its Affiliates.
(c) Sepracor hereby grants to GSK, a non-exclusive license throughout the GSK Territory under the Sepracor Technology to undertake Developmental Studies and any other activities specified in the Development Plan with respect to the Product and consistent with this Agreement.
(d) Sepracor hereby grants to GSK an exclusive license to use the Product Trademarks and Other Trademarks in the GSK Territory on the terms of Section 13.4.
2.2. Activities Outside the GSK Territory.
(a) GSK agrees that neither it, nor any of its Affiliates or Sublicensees, will (i) export the Product outside of the GSK Territory, or (ii) sell or provide the Product to any Third
CONFIDENTIAL
Party if GSK and/or its relevant Affiliate or Sublicensee believes, or it would be reasonable to assume, that the Product sold or provided to such Third Party will be exported for use outside the GSK Territory.
(b) Sepracor agrees that neither it, nor any of its Affiliates, will (i) export the Product within the GSK Territory (other than to GSK, its Affiliates, or Sublicensees), or (ii) sell or provide the Product to any Third Party if Sepracor or its relevant Affiliate or Third Party sub-licensee believes, or it would be reasonable to assume, that the Product sold or provided to such Third Party will be imported for use in the GSK Territory.
2.3. No Other Rights. Except for the rights and licenses expressly granted in this Agreement, Sepracor retains all rights under its intellectual property and no additional rights shall be deemed granted to GSK by implication, estoppel or otherwise. For the avoidance of doubt, Sepracor specifically retains, without limitation, its rights to (i) develop, manufacture, license, use, store, import, export, distribute, promote, market, offer for sale and sell the Excluded Products in the GSK Territory, (ii) develop, manufacture, license, use, store, import, export, distribute, promote, market, offer for sale and sell within the GSK Territory products which are not specifically covered under this Agreement; and (iii) manufacture or have manufactured Product within the GSK Territory for use or sale outside of the GSK Territory.
ARTICLE III.
GOVERNANCE
3.1. Steering Committee. The Parties shall establish a joint steering committee (the “JSC”) consisting of an equal number of representatives of senior management from each Party, each such representative having the authority to act on behalf of the Party such individual represents, the exact number of which shall be as the Parties may agree, from time to time. Initially, the JSC shall consist of six (6) individuals; three (3) of whom shall be nominated by Sepracor; and three (3) of whom shall be nominated by GSK. Any member of the JSC may designate a substitute to attend and perform the functions of that member at any meeting of the JSC. Each Party may, with the consent of the other Party, such consent not to be unreasonably withheld or delayed, invite non-member, non-voting representatives of such Party to attend meetings of the JSC. Each Party shall on an alternate year basis designate the chairperson and the other Party shall designate the secretary of the JSC. The initial chairperson shall be designated by GSK; and the initial secretary shall be designated by Sepracor. The JSC shall perform the following responsibilities:
(a) oversee the overall strategy for the development (if any) and commercialization of the Product in the GSK Territory;
(b) facilitate communication between the two Parties and provide a forum to review any development, regulatory, manufacturing and commercialization matters pertaining to the Product;
CONFIDENTIAL
(c) provide a forum for communication of GSK’s activities in the GSK Territory, and Sepracor’s and Third Party licensees’ activities in the Sepracor Territory, with respect to the Product;
(d) review any Regulatory Authority requirements for Required Studies;
(e) undertake a bi-annual (two times per year) review and comparison of the status of the Development Plan (if any) and Commercialization Plan, including, without limitation the applicable timelines, and provide direction to the conduct of the Development Plan and Commercialization Plan, as necessary;
(f) review and approve the proposed Development Plan and any fundamental amendments or modifications thereto;
(g) review any Sepracor Studies which are planned to be conducted in the GSK Territory, provided that the extent of such review shall be limited to a brief description of the Sepracor Study, including the location(s) and primary purpose of such study, and shall not include a review of the protocol or investigator brochure for such study;
(h) review and approve the first proposed Core Campaign Product Strategy and any subsequent versions which are a material change of strategy or visuals, proposed by GSK for the GSK Territory and discuss any potential benefits from synergies of such activities in the GSK Territory with efforts in the Sepracor Territory;
(i) review and approve any additional brand names and Other Trademarks, subject to Article XIII;
(j) coordinate, oversee and delegate the activities of any Subcommittees established pursuant to Section 3.2 of this Agreement;
(k) in accordance with Section 3.2, resolve disputes, disagreements and deadlocks unresolved by any Subcommittees established pursuant to Section 3.2 of this Agreement; and
(l) perform such other responsibilities as may be assigned to the JSC pursuant to this Agreement or as may be mutually agreed upon by the Parties from time to time.
3.2. Subcommittees. From time to time, the JSC may establish subcommittees to oversee particular projects or activities within the scope of authority of the JSC, as it deems necessary or advisable (each, a “Subcommittee”). Each Subcommittee shall consist of such number of representatives of each Party as the JSC determines is appropriate from time to time. Each Subcommittee shall meet with such frequency as the JSC shall determine. All decisions of each Subcommittee shall be made by unanimous vote or written consent, with Sepracor and GSK each having, collectively, one vote in all decisions. If, with respect to a matter that is subject to a Subcommittee’s decision-making authority, the Subcommittee cannot reach unanimity, the
CONFIDENTIAL
matter shall be referred to the JSC, which shall resolve such matter in accordance with Section 3.4. The Parties envisage that the JSC will establish Subcommittees shortly after the Effective Date to oversee each of the following areas: (i) regulatory matters; (ii) manage transition activities; (iii) advise on optimum commercial strategies; and (iv) supply matters. If one Party expressly requires that a Subcommittee be appointed, then it will be promptly established by the JSC.
3.3. Meetings. All JSC and Subcommittee meetings shall be as often as the members may determine, but in any event JSC meetings shall occur not less than twice per calendar year. Such meetings may be held in person, or any means of telecommunications or video conference, as the members deem necessary or appropriate; provided, however, that at least one JSC meeting per year shall be held in person and the location of such in person meeting shall alternate between Sepracor’s and GSK’s offices. A quorum for JSC meetings shall be four (4) members, with at least two (2) members from each Party.
3.4. Decision-making of Joint Steering Committee.
(a) The JSC may make decisions with respect to any subject matter that is subject to the JSC’s decision-making authority or within the purview of any other Subcommittee organized as part of this Agreement. Except as expressly provided in this Agreement, all decisions of the JSC shall be made by unanimous vote or written consent, with Sepracor and GSK each having, collectively, one vote in all decisions. The JSC shall use reasonable efforts to resolve any disputes concerning the matters within its roles and functions or otherwise referred to it.
(b) If, with respect to a matter that is subject to the JSC’s decision-making or oversight authority, the JSC cannot reach consensus within fifteen (15) days after it has met and attempted to reach such consensus or the Parties cannot reach consensus on whether the JSC has decision-making authority regarding a matter within fifteen (15) days after such matter was first raised by either Party, the dispute in question shall be referred to the Chief Executive Officer of Sepracor and the President, Pharmaceuticals Europe of GSK (collectively, the “Senior Executives”), for resolution. The Senior Executives shall use reasonable efforts to resolve the matter referred to them with fifteen (15) days of such referral.
(c) If the Senior Executives cannot resolve the matter in accordance with Section 3.4(b), matters in dispute shall be finally determined as follows:
(i) all regulatory matters shall be finally determined by the President, Pharmaceuticals Europe of GSK, except as to matters otherwise more specifically discussed in Sections 4.2(b) and (c), which shall be finally determined by the Chief Executive Officer of Sepracor;
(ii) all commercialization issues (except as to matters otherwise more specifically discussed in Sections 5.7 and 6.2) shall be finally determined by the President, Pharmaceuticals Europe of GSK;
CONFIDENTIAL
(iii) all matters regarding the Developmental Studies to be conducted by GSK (except as to matters otherwise more specifically discussed in Section 5.7) shall be finally determined by the President, Pharmaceuticals Europe of GSK; and
(iv) all matters regarding Sepracor Studies shall be finally determined by the Chief Executive Officer of Sepracor.
(d) For all purposes under this Agreement, any decision made pursuant to this Section 3.4 shall be deemed to be the decision of the JSC.
(e) Neither Party will exercise its right to finally resolve a dispute in accordance with this Section 3.4 in a manner that excuses such Party from any of its obligations specifically enumerated under this Agreement.
(f) Notwithstanding this Section 3.4, any dispute regarding the interpretation of this Agreement or any alleged breach of this Agreement will be resolved in accordance with the terms of Article 18.
3.5. Minutes. Minutes for each of the JSC meetings shall be drafted by the secretary of the meeting and sent to the chairperson of the applicable committee for comment promptly after each such meeting (but in no event more than thirty (30) days). All actions noted in the minutes are to be reviewed and approved at subsequent meetings of the JSC; provided, that if the Parties cannot agree as to the content of the minutes, such minutes will be finalized to reflect such disagreement.
3.6. Expenses. Each Party shall bear all its own costs, including expenses incurred by the members nominated by it in connection with their activities as members of the JSC.
3.7. Alliance Managers. Promptly after the Effective Date, each Party shall appoint an individual to act as the alliance manager for such Party (the “Alliance Manager”). Each Alliance Manager who is not otherwise a member of the JSC shall thereafter be permitted to attend meetings of the JSC. The Alliance Managers shall be the primary contact for the Parties regarding the activities contemplated by this Agreement and shall facilitate all such activities hereunder. Each Party may replace its Alliance Manager with an alternative representative at any time with prior written notice to the other Party. The Alliance Managers shall not, in any manner, take over the role of the JSC and shall not have any rights, powers or discretion except as expressly granted to the Alliance Managers hereunder. In no event shall the Alliance Managers have any power to modify or amend this Agreement
CONFIDENTIAL
ARTICLE IV.
TRANSITION ACTIVITIES
4.1. Current Status; General. Prior to the Effective Date, Sepracor (alone or with Third Parties) has conducted pre-clinical and clinical studies of the Product, and on 23rd July 2007 submitted a MAA for the Product to the EMEA for review under the centralized procedure.
4.2. Regulatory Matters.
(a) During the Transition Period, Sepracor shall retain all right, title and interest in and to all Marketing Authorization Applications for the Product in the GSK Territory, subject to the licenses granted to GSK pursuant to this Agreement.
(b) During the Transition Period, Sepracor shall support the MAA filed with the EMEA on 23rd July 2007 and use its Commercially Reasonable Efforts to obtain approval for the MAA from the EMEA for the Product with labeling consistent with, or less restrictive than, the Target Label. Sepracor shall be responsible for liaising with and managing all interactions with the EMEA in relation to the Product in connection with such MAA. Sepracor shall provide GSK with reasonable advance notice of any meetings with the EMEA (including any oral hearings), if practicable, and GSK shall have the right, but not the obligation, to attend and participate in such meetings where legally permissible. In addition, Sepracor shall promptly provide GSK with copies of all material correspondence from the EMEA (including any assessment reports) related to the Product in the GSK Territory, and shall provide GSK with draft copies of all material correspondence intended for the EMEA (including any response documents) for review and agrees to consider in good faith any prompt comments GSK may have with respect to any such regulatory filings or interactions.
(c) During the Transition Period, Sepracor shall remain responsible for liaising with and managing all interactions with other Regulatory Authorities in relation to the Product in connection with a MAA for any country in the GSK Territory, except for those countries where the Parties have mutually agreed to earlier transfer responsibility to GSK. Sepracor shall not however initiate the filing of any new MAA, or initiate any interaction, with any Regulatory Authorities (other than the EMEA) within the GSK Territory related to the Product without the prior approval of GSK. Sepracor shall provide GSK with reasonable advance notice of any meetings with any such Regulatory Authority (including any oral hearings) about the Product, if practicable, and GSK shall have the right, but not the obligation, to attend and participate in such meetings where legally permissible. In addition, Sepracor shall promptly provide GSK with copies of all material correspondence from any such Regulatory Authorities (including any assessment reports) related to the Product in the GSK Territory, and shall provide GSK with draft copies of all material correspondence intended for any such Regulatory Authorities (including any response documents) for review and agrees to consider in good faith any comments GSK may have with respect to any such regulatory filings or interactions with respect to the Product. Where the Parties have agreed to transfer responsibility
CONFIDENTIAL
to GSK for any country in the GSK Territory before the expiration or termination of the Transition Period, Sections 4.2(d), 5.1 and 5.14 shall apply mutatis mutandis.
(d) Subject to obtaining all required consents or approvals from all relevant Regulatory Authorities, as soon as reasonably practicable following the expiration or termination of the Transition Period, Sepracor shall transfer to GSK all Marketing Approvals and/or XXXx for the Product in the GSK Territory (except for DMFs), with Sepracor to invoice GSK for one-half of any out-of-pocket costs for so doing. In addition, Sepracor agrees to promptly execute, acknowledge and deliver such further instruments, and to do all such other reasonable acts, as may be necessary in order to carry out the transfer of such Marketing Approvals and/or XXXx, as the case may be related to the Product in the GSK Territory.
(e) Within [**] following the Effective Date, GSK and Sepracor shall provide for review by the JSC an outline showing in reasonable detail GSK’s provisional plans for filing Marketing Authorization Applications and obtaining Marketing Approvals in the GSK Territory, with a particular focus on the overall strategy and timeline of material submissions and filings (if any) during the Transition Period and how the various regulatory interactions will be managed during the Transition Period.
4.3. Required Studies (EMEA). In the event that the EMEA requires or requests in writing that Required Studies (other than those already initiated by Sepracor as of the Effective Date) to be performed as a condition of, or in connection with, granting Marketing Approval, GSK shall have the right, at its absolute discretion, to perform or have performed such Required Studies at GSK’s expense. If GSK decides to perform or have performed such Required Studies, the JSC will review and discuss which of the Parties should undertake such Required Studies. Sepracor will provide all reasonable incidental assistance to GSK and/or its Affiliates if GSK wishes to undertake the Required Studies itself, or via its Affiliates or any sub-contractor. Sepracor shall provide such assistance free of charge, unless otherwise agreed in writing by GSK. In the event that GSK notifies Sepracor that it does not desire to incur such expense, at any time after such notification Sepracor may, but shall not be obligated to, terminate the Agreement with respect to the European Union (but not the rest of the GSK Territory) upon sixty (60) days written notice. For the avoidance of doubt, a decision not to pursue such Marketing Approval by GSK shall not be deemed a failure to exercise its Commercially Reasonable Efforts.
In the event that Sepracor believes that the use of the results of any Required Study could be of material benefit to Sepracor in the Sepracor Territory, for example, if the Required Study were the development of a [**] of the Product, the Parties shall [**], taking into account the costs of any assistance which may have been provided by Sepracor under this Section.
4.4. Reporting: Adverse Drug Reactions. During the Transition Period, Sepracor shall, in accordance with Section 5.14, be responsible for filing all reports required to be filed in order to maintain any IND, MAA and/or any Marketing Approvals filed or granted for the Product in the GSK Territory, including reporting of adverse drug experiences.
CONFIDENTIAL
ARTICLE V.
REGULATORY MATTERS AND DEVELOPMENT ACTIVITIES
5.1. Regulatory Matters.
(a) Following the Transition Period and for the Term of this Agreement, GSK shall retain all right, title and interest in and to all Marketing Authorization Applications and Marketing Approvals for the Product in the GSK Territory, subject to the licenses granted to Sepracor pursuant to Section 5.12 and the assignment and transfer of such Marketing Authorization Applications and Marketing Approvals pursuant to Section 15.2(b).
(b) Upon expiration or termination of the Transition Period, GSK shall have sole responsibility for liaising with and managing all interactions with Regulatory Authorities in relation to the Product in the GSK Territory in connection with any and all regulatory filings. GSK shall provide Sepracor with reasonable advance written notice of any meetings with the Regulatory Authorities related to the Product in Major Markets of the GSK Territory and Sepracor shall have the right, but not the obligation, to attend and participate in such meetings in the Major Markets where legally permissible. Notwithstanding the foregoing, GSK shall provide Sepracor with a copy of any regulatory submission and other substantive written material to be sent to any Regulatory Authority in the GSK Territory for review and agrees to consider in good faith any prompt comments Sepracor may have with respect to any such regulatory filings or interactions.
(c) Sepracor shall provide GSK with all reasonable assistance, in a timely manner, to support GSK’s activities in seeking and maintaining Market Approval in the GSK Territory. Without limiting the above, Sepracor accepts that such assistance may extend to obtaining Certificates of Pharmaceutical Product on behalf of GSK.
(d) Following the Transition Period GSK shall report to the JSC the overall strategy and timeline of all material submissions and filings with any Regulatory Authority relating to the Product in the GSK Territory.
(e) Each Party shall promptly notify the other Party in writing of any material changes or material issues that may arise in connection with any IND, MAA and/or any Marketing Approvals in any country within their respective territories.
(f) Sepracor acknowledges that in order for Marketing Approval to be obtained in many countries within the GSK Territory but outside of the European Union, it will be necessary for GSK to obtain a Certificate of Pharmaceutical Product from a suitable reference country, either in the GSK Territory or in the Sepracor Territory. Sepracor agrees to provide GSK will all reasonable assistance in obtaining such Certificate of Pharmaceutical Product, and without limiting the foregoing, agrees in particular to:
CONFIDENTIAL
(i) work with GSK to establish a supply route for the Product which is capable of supporting the issue of a Certificate of Pharmaceutical Product from a suitable reference country;
(ii) generate, if not already available, at GSK’s expense, 30/75 stability data for the Product using the supply route established under Section 5.1(f)(i) above;
(iii) generate sufficient number of Product samples, at GSK’s expense, to support GSK’s activities in seeking and maintaining Market Approval in the GSK Territory.
(g) Sepracor further acknowledges that until a supply route has been established under Section 5.1(f)(i), GSK will be unable to provide detailed regulatory or commercialization plans, where required under this Agreement, in respect of such countries where a Certificate of Pharmaceutical Product is required.
5.2. Regulatory Plan. Within ninety (90) days following the expiry or termination of the Transition Period, GSK shall provide for review by the JSC an outline plan showing GSK’s plans for filing Marketing Authorization Applications and obtaining Market Approval in the GSK Territory. The regulatory plan shall include a description of material regulatory activities to be undertaken during the following [**] years, including but not limited to, planned tasks and timelines for the conduct of the regulatory activities with respect to all matters necessary to obtain Marketing Approval for the Product in each Major Market of the GSK Territory.
5.3. Required Studies (Non-EMEA). In the event that any Regulatory Authority in the GSK Territory (other than the EMEA) requires or requests in writing that Required Studies (other than those already initiated by Sepracor as of the Effective Date) be performed as a condition of, or in connection with, granting Marketing Approval, [**] whether or not to conduct such Required Studies and continue to [**] in such country. If [**] shall undertake such Required Studies [**]. In the event that [**] does not [**], in relation to countries other than those within the European Union or for which a date for accession to the European Union has been agreed, Sepracor may, but shall not be obligated to, terminate the Agreement with respect to the particular country or countries where such Regulatory Authority has required such clinical trials and [**], and the Agreement shall remain in full force and effect in the rest of the GSK Territory. For the avoidance of doubt, a [**] shall not be deemed a failure to exercise its Commercially Reasonable Efforts.
In the event that [**] to undertake such Required Study, and Sepracor believes that the use of the results of any Required Study could be of material benefit to Sepracor in the Sepracor Territory, for example, if the Required Study were the development of a [**] of the Product, the Parties [**].
5.4. Additional Development Activities of GSK. In addition to the studies permitted in Sections 4.3 and 5.3, and in accordance with the then applicable approved Development Plan, GSK shall have the right, but not the obligation, to undertake, at GSK’s sole
CONFIDENTIAL
expense, Commercialization Studies in the GSK Territory in support of GSK’s commercialization of the Product. GSK shall inform Sepracor pursuant to Section 5.6 of its intention to pursue such Commercialization Studies prior to their initiation.
5.5. Investigator Sponsored Clinical Study. For the avoidance of doubt, GSK shall be free, at GSK’s sole expense, to provide financial or in-kind support to clinical studies of the Product that are sponsored and conducted by a Third Party and undertaken in the GSK Territory (“Investigator Sponsored Clinical Studies”). GSK shall inform Sepracor pursuant to Section 5.6 of its intention to provide financial or in-kind support to such Investigator Sponsored Clinical Studies prior to providing such support.
5.6. Development Plan. In the event that GSK plans during the Term of this Agreement to undertake any Required Studies or Commercialization Studies, or to support any Investigator Sponsored Clinical Studies in the XXX Xxxxxxxxx, XXX shall generate a Development Plan which shall be presented promptly to the JSC for review to ensure that the Development Plan takes into account and is consistent with the global branding strategy for the Product. The Development Plan shall be supplemented, modified and updated regularly by GSK as and when additional relevant data and information become available, but in any event not less frequently than annually. Required Studies, Commercialization Studies and support for Investigator Sponsored Clinical Studies shall not commence until such studies have been presented to the JSC for review and written notice of the studies sent to Sepracor. GSK shall keep the JSC advised on a timely basis of the progress of each such study.
5.7. Sepracor’s Review and Final Approval. Notwithstanding anything to the contrary contained in this Agreement, in the event that Sepracor in good faith believes that any Development Studies GSK wishes to undertake or sponsor pursuant to Sections 5.3 or 5.4, or any Investigator Sponsored Clinical Studies GSK wishes to support pursuant to Section 5.5 has a [**], Sepracor may provide written notice to GSK within [**] of such studies being presented to the JSC, requesting that GSK shall not perform such Developmental Studies and/or shall not support such Investigator Sponsored Clinical Studies.
(a) If such notice is received by GSK within the [**] period, GSK shall not commence such Developmental Study or support such Investigator Sponsored Clinical Study until mutual agreement or resolution of any dispute has been achieved in respect of the performance of such study.
(b) If GSK disputes the assessment of the [**], GSK may refer the dispute to the Senior Executives. The Senior Executives shall negotiate in good faith to resolve the dispute within [**], or such longer period as mutually agreed. If the Senior Executives are unable to resolve the dispute within such time period, [**].
(c) For the avoidance of doubt, if Sepracor does not provide such written notice with the above stated period, Sepracor’s approval will be deemed to have been received.
CONFIDENTIAL
5.8. Development Activities of Sepracor in the GSK Territory. Sepracor shall have the right, but not the obligation, to undertake future development within the GSK Territory in support of the commercialization of Product in the Sepracor Territory and the right, but not the obligation, to undertake future development within the GSK Territory of Excluded Products. However, with respect to Sepracor Studies, Sepracor shall generate a Sepracor Development Plan (the “Sepracor Development Plan”) which shall be presented promptly to the JSC for review to ensure that the Sepracor Development Plan takes into account, and is not inconsistent with, the branding strategy for the Product in the GSK Territory. The Sepracor Development Plan shall include reasonably detailed information regarding all such activities. It is acknowledged however that in respect of Sepracor Studies not directly related to the Product, the level of information that will be required to be disclosed to enable appropriate review may be limited (except in respect of matters relating to safety).
5.9. Conduct of Activities. The Parties shall carry out all pre-clinical and clinical studies related to the Product that they respectively undertake in the GSK Territory in compliance with all applicable laws, rules and regulations and in accordance with good scientific and clinical practices, applicable under the laws and regulations of the relevant country in the GSK Territory.
5.10. Drug Master File.
(a) Responsibility for Maintaining. Notwithstanding Section 5.1(a), Sepracor shall be responsible for filing and maintaining Drug Master Files (“DMF”) relating to the Product and Active Ingredient contained in the Product, where such DMF is either required by a Regulatory Authority or it is determined by the JSC that DMF submission is preferable to incorporation of the DMF information in the applicable MAA. Sepracor shall file and maintain such DMFs in its own name, and/or in the name of its relevant suppliers, and shall permit GSK, its Affiliates and Sublicensees, to cross-reference such DMFs in their regulatory filings for the Product in the GSK Territory.
(b) Costs. Responsibility for the costs of filing and maintaining such DMFs shall be Sepracor’s.
(c) Closed Portions. Notwithstanding any other provisions of this Agreement, the definitions of “Data” and “Sepracor Know-How” shall not include the closed portions of any such DMF, nor any information contained in any such portions of the DMF and thus, notwithstanding any other provision of this Agreement, Sepracor shall have no obligation to disclose to GSK or any of its Affiliates or Sublicensees the closed portions of such DMFs or any information contained therein. For the avoidance of doubt, Regulatory Authorities in the GSK Territory shall have the right to access the entire DMF, including any closed portions.
5.11. Exchange of Data and Know-How.
(a) By Sepracor. Subject to the terms and conditions of this Agreement, Sepracor shall promptly make available to GSK all relevant Sepracor Know-How relating to the
CONFIDENTIAL
Product, including all Data from any and all clinical trials and preclinical studies for the Product that are Controlled by Sepracor as of the Effective Date or that will be obtained by Sepracor thereafter, subject in each case to Section 5.10. Sepracor shall also use its Commercially Reasonable Efforts to provide to GSK any Third Party manufacturer’s know-how which is necessary for GSK to obtain Marketing Approval in the GSK Territory,
(b) New Data. Notwithstanding Sections 5.10, 5.11(a) and 5.12, in the event that GSK requires information relating to the Bulk Tablets in addition to that provided by or otherwise readily available to Sepracor to support GSK’s regulatory filings for Product in the GSK Territory, Sepracor shall conduct, at GSK’s expense, such work as is necessary to produce the necessary chemistry, manufacturing and controls (“CMC”) information or other regulatory documentation reasonably required to support such regulatory filings (provided that GSK shall be responsible for, at its expense, any additional clinical trial work required for such filings). The obligation of Sepracor to provide such CMC data or other regulatory documentation reasonably required to support such regulatory filings will include, without limitation, CMC information or other regulatory documentation reasonably required to support such regulatory filings beyond what was required for the U.S.A. regulatory approval, to support initial as well as filed regulatory submissions for Product in the GSK Territory, support on an ongoing basis to deal with regulatory questions, provision of on-going stability data, batch analysis, etc. for license renewals, CMC data for variations, any response to questions relating to renewals and variations, as well as any other data required to meet any regulatory inspection requirements. For the avoidance of doubt, Sepracor’s obligations do not extend to questions regarding finishing operations performed by GSK.
(c) Provision of Data to JSC. Upon request by the JSC, GSK and Sepracor shall promptly provide the JSC with summaries in reasonable detail of all Data generated or obtained in the course of the performance of activities under the Development Plan or the Sepracor Development Plan respectively. For the avoidance of doubt, this obligation shall not extend to provision of data not related to the Product and only relating to Excluded Products.
5.12. Sharing of Regulatory Filings. Without limiting Section 5.11 above, and without prejudice to the terms of the Supply Agreement, GSK and Sepracor shall permit the other to access, and shall provide the other Party with sufficient rights to reference and use in association with exercising its rights and performing its obligations under this Agreement, all of such Party’s, its Affiliates’ and Sublicensees’ Data, regulatory filings and material regulatory communications associated with any submissions of INDs, XXXx, Marketing Approvals or other approvals for the Product in the GSK Territory and in the Sepracor Territory, respectively. It is understood that such rights and access shall be limited respectively to Sepracor’s, its Affiliate’s and sublicensees’ commercialization of the Product in the Sepracor Territory and to GSK’s, its Affiliate’s and Sublicensee’s commercialization of the Product in the GSK Territory.
5.13. Regulatory Co-operation. Without limiting Section 5.11 above, in the event that either Party receives enquiries from a Regulatory Authority in their respective territory, regarding filings or communications made, or activities being undertaken in the other Party’s
CONFIDENTIAL
respective territory, the other Party shall provide all reasonable co-operation to enable the original Party to respond to the Regulatory Authority’s enquiries.
5.14. Reporting: Adverse Drug Reactions.
(a) Sepracor and GSK shall be responsible for filing all reports required to be filed in order to maintain any IND, MAA and/or any Marketing Approvals granted for the Product in the Sepracor Territory or the GSK Territory, respectively, including reporting of adverse drug experiences; provided, however, that during the Transition Period, Sepracor shall be responsible for such filings in the GSK Territory.
(b) The Parties shall be responsible to ensure that their respective Affiliates and sublicensees comply with all such reporting obligations.
(c) The Parties will exchange all pharmacovigilance relevant information pursuant to a separate pharmacovigilance agreement to be executed within ninety (90) days of the Effective Date (the “Pharmacovigilance Agreement”). The standards set forth in the Pharmacovigilance Agreement shall be no less stringent than those set forth in the ICH Guidelines and shall provide, among other things, that:
(i) The Parties will comply with regulatory requirements for the reporting of safety data in accordance with standards stipulated in the ICH Guidelines, and all applicable regulatory and legal requirements regarding the management of safety data;
(ii) The Parties will exchange relevant safety data within appropriate timeframes and in an appropriate format to enable them to meet both expedited and periodic local and international regulatory reporting requirements and to facilitate appropriate safety reviews; and
(iii) Sepracor shall be the holder of the world-wide safety database for all adverse events.
(d) Furthermore, Sepracor commits to the establishment of pharmacovigilance systems in compliance with applicable regulatory requirements and to demonstrate comprehensively those systems, particularly their system for producing PSURs, to GSK prior to the end of the Transition Period. In the event that GSK has any reasonable concerns after such demonstration, GSK and Sepracor will co-operate in good faith to resolve such concerns.
(e) Prior to the end of the Transition Period, Sepracor shall establish a separate pharmacovigilance agreement with each Third Party to which Sepracor has granted rights to promote, market and sell the Product in the Sepracor Territory, or part thereof. In the event that after the Effective Date, Sepracor grants to any Third Party rights to promote, market and sell the Product in the Sepracor Territory, or any part thereof, Sepracor shall enter into a separate pharmacovigilance agreement in connection with such grant. The standards set forth in
CONFIDENTIAL
such pharmacovigilance agreement(s) shall be not less stringent than as provided for under (c) above.
5.15 Marketing Approval Challenge. In the event that Sepracor or GSK becomes aware of an actual or threatened challenge to the data exclusivity rights relating to any Marketing Approval within the GSK Territory, that Party shall promptly notify the other Party in writing. The Parties shall meet to discuss a coordinated response to such challenge prior to any action being taken. Neither Party shall be required by the other Party to take any action in relation to such challenge, which in its absolute discretion it does not want to take. Each Party shall be entitled, without obligation, to independently initiate proceedings or take other appropriate action in relation to such challenge at its own expense. The Party conducting such action shall have full control over its conduct, including settlement thereof; provided, however, that the Party conducting such action may not settle any such action, or make any admissions or assert any position in such action, in a manner that would materially adversely affect the rights or interests of the other Party whether within or outside the jurisdiction in which the action is brought, without the prior written consent of the other Party, which consent shall not be unreasonably withheld or delayed.
ARTICLE
VI.
COMMERCIALIZATION, MARKETING AND PROMOTION
6.1. Commercialization Plan. The parties understand that the level of specificity of the Commercialization Plan will vary according to the stage of development of the Product relative to Marketing Approval and/or determination of pricing in the Major Markets. However, the following shall apply to the development, review and approval of such Commercialization Plan:
(a) On an annual basis, GSK shall prepare and deliver to the JSC, for its review, a proposed plan for the marketing, sale, advertising, and sales activities (including pre-launch market development and market research) for the Product in the GSK Territory. For the Major Markets, the plan will outline in as much detail as practicable given the stage of commercialization of the Product annual [**]. (The first such plan being the “Initial Commercialization Plan” and such later plans, including any amendments to the Initial Commercialization Plan, as amended from time to time, herein after referred to as the “Commercialization Plan”). The Initial Commercialization Plan shall be delivered on or before [**].
(b) The Commercialization Plan shall be supplemented, modified and updated regularly by GSK as and when additional relevant data and information become available, but in any event not less frequently than annually. Any fundamental modifications made to the Commercialization Plan shall be delivered to the JSC for review promptly following such modifications, even when such may occur between regularly scheduled meetings.
CONFIDENTIAL
6.2. Marketing and Advertising Campaign. GSK shall provide the JSC with a detailed overview of the first proposed Core Campaign Product Strategy and any subsequent versions which are a material change of strategy or visuals (and any local core marketing materials that fall outside the Core Campaign Product Strategies) to be used in the GSK Territory, which the JSC shall review to ensure that the Core Campaign Product Strategy shall be consistent with the global branding strategy for the Product. Notwithstanding anything to the contrary contained in this Agreement, in the event that Sepracor in good faith believes that any part of the proposed Core Campaign Product Strategy has a reasonable likelihood of materially adversely affecting the volume of net sales in the Sepracor Territory, Sepracor may provide written notice to GSK within [**] of such Core Campaign Product Strategy being presented to the JSC requesting that such part of the Core Campaign Product Strategy not be implemented.
(a) If such notice is received by GSK within the [**] period, GSK shall not implement such part of the core marketing and advertising campaign until mutual agreement or resolution of any dispute has been achieved in respect of the use of the proposed core marketing and advertising campaign.
(b) If GSK disputes the assessment of the reasonable likelihood of material adverse effect, GSK may refer the dispute to the Senior Executives. The Senior Executives shall negotiate in good faith to resolve the dispute within [**]. If the Senior Executives are unable to resolve the dispute within such time period, Sepracor’s decision shall be binding.
(c) For the avoidance of doubt, if Sepracor does not provide such written notice with the above stated period, Sepracor’s approval will be deemed to have been received.
6.3. Responsibilities of GSK under the Commercialization Plan. Other than manufacturing of the Product, GSK shall have the exclusive right to engage in, and shall be solely responsible for, all activities set forth in the Commercialization Plan and [**]. As part of the Commercialization Plan, GSK shall:
(a) use its Commercially Reasonable Efforts to commercialize the Product in the GSK Territory, including seeking approval from relevant Regulatory Authorities regarding price and reimbursement of the Product in the GSK Territory;
(b) use its Commercially Reasonable Efforts to perform pre-commercialization analysis, planning, market preparation and related marketing activities in the GSK Territory;
(c) use its Commercially Reasonable Efforts to carry out the distribution, marketing and sales of the Product in the GSK Territory;
(d) incur at least an aggregate of [**] Dollars ($[**]) in support of pre-launch marketing efforts allocated amongst all the countries in the GSK Territory;
CONFIDENTIAL
(e) conduct the Commercialization Plan in compliance with all applicable laws;
(f) provide suitable storage and handling in accordance with the labeling and specifications for the Product and other appropriate facilities and services as needed for the storage and continuous sale and distribution of the Product throughout the GSK Territory;
(g) provide all medical information services for the Product in the GSK Territory, which may include, telephone hotlines, informational fax programs, mail response programs;
(h) administer and pay any chargebacks, rebates, administrative service fees and other discounts and fees with respect to the Product in the GSK Territory which from time to time GSK may agree to give or may be required under applicable laws to give to payors or purchasers of the Product;
(i) use its Commercially Reasonable Efforts to adequately train sales representatives;
(j) use its Commercial Reasonable Efforts to undertake an analysis of the intellectual property position in any country of the GSK Territory where a launch of the Product is anticipated, and where deemed necessary by GSK, develop Other Trademarks for review and approval by the JSC;
(k) use Commercially Reasonable Efforts to conduct its commercialization of the Product in accordance with the Commercialization Plan ; and
(l) [**] after the end of each Calendar Year, furnish Sepracor with reasonably detailed summary written reports on all activities conducted by GSK under the Commercialization Plan during such calendar year. Information shall be provided on a country by country basis for the Major Markets.
6.4. Failure to Launch.
(a) For the avoidance of doubt and subject to the other terms in this Section 6.4, the Parties hereby accept that the failure to launch the Product in any country or countries shall not, per se, be deemed a breach of GSK’s obligations to use Commercially Reasonable Efforts to commercialize the Product. Where GSK’s decision not to launch in any specific country is founded on GSK’s good faith belief that the Product is not commercially viable in such country, for example because of pricing and reimbursement concerns or that the launch of the Product in such country would adversely affect the amount of Net Sales that would be achieved in the XXX Xxxxxxxxx, XXX shall not be required to and Sepracor shall not have the right to terminate this Agreement in such country, except as described in Section 6.4(b). However, for the avoidance of doubt: (i) GSK’s good faith belief must be supported by actual interactions with Regulatory Authorities or other good faith objective evidence which supports
CONFIDENTIAL
GSK’s conclusions; (ii) GSK must share its financial assessment, in reasonable detail, setting forth why the Product is not in good faith deemed commercially viable; and (iii) the JSC is to be notified and have the opportunity to review any such decision not to launch the Product in a country.
(b) Nothwithstanding anything else herein, where (i) the GSK decisions in Section 6.4(a) at any time combine so that GSK has decided that it will not launch in [**], or (ii) GSK has failed to launch in [**] following EMEA approval, Sepracor has the right to terminate this Agreement, on a country-by-country basis, in [**] by giving GSK sixty (60) days written notice to that effect at any time thereafter, provided that GSK has not in the interim, prior to receipt of notice of termination from Sepracor, notified Sepracor that it plans to launch in [**].
6.5. Price and Reimbursement. GSK shall be solely responsible for any decisions and negotiations with relevant Regulatory Authorities regarding price and reimbursement of the Product in the GSK Territory. Sepracor shall be fully and contemporaneously updated of on all efforts by GSK with respect to pricing and reimbursement matters provided that the disclosure of such information is for information purposes only.
6.6. Booking Sales. GSK will book all sales for the Product in the GSK Territory.
6.7. Sepracor Support. Sepracor shall provide GSK with reasonable assistance, in a timely manner, to support GSK’s activities in commercializing the Product in the GSK Territory, including but not limited to provision of a representative set of Sepracor Materials (as defined below). Sepracor Materials shall mean copies of advertising, promotional, educational and communication materials, produced by Sepracor for marketing, advertising and promotion of Product for distribution to Third Parties (including medical professionals) and to Sepracor’s sales forces in the United States, together with programs related to patient education, physician education and disease management programs.
ARTICLE
VII.
PAYMENTS
7.1. Initial License Fee. In consideration for the licenses granted hereunder under Sepracor Patents and Sepracor Know-How, GSK shall pay to Sepracor an initial license fee in the amount of Twenty Million US Dollars ($20,000,000) within five (5) Business Days following GSK’s receipt of an invoice for such license fee in accordance with the payment provisions of Article VIII. Sepracor intends to deliver to GSK within five (5) Business Days of the Effective Date such invoice for the license fee. The initial license fee set forth in this Section shall not be refundable or creditable against any future milestone payments, royalties or other payments by GSK to Sepracor under this Agreement.
CONFIDENTIAL
7.2. Milestone Payments.
(a) Milestone Payments. In further consideration for the licenses granted hereunder under Sepracor Patents and Sepracor Know-How, GSK shall pay to Sepracor the milestone payments set out below following the first achievement by GSK, or any of its Affiliates or Sublicensees, individually or in the aggregate, of the corresponding milestone, in accordance with this Section 7.2 and the payment provisions in Article VIII:
|
Milestone Events |
|
Milestone |
|
|
|
Centralized EMEA approval with new active substance status and Target Label |
|
$ |
[**] |
|
|
Positive [**] |
|
$ |
[**] |
|
|
Approved reimbursement price of €[**] or more per [**]mg tablet in France |
|
$ |
[**] |
|
|
Approved reimbursement price of €[**] or more per [**]mg tablet in Germany |
|
$ |
[**] |
|
|
Sales Threshold Milestones |
|
Milestone |
|
|
|
First achievement of Calendar Year Net Sales of €[**] (2) |
|
$ |
[**] |
|
|
First achievement of Calendar Year Net Sales of €[**] |
|
$ |
[**] |
|
|
First achievement of Calendar Year Net Sales of €[**] |
|
$ |
[**] |
|
|
Total Potential Milestones |
|
Total |
|
|
|
Potential Milestone Events plus Potential Sales Threshold Milestones |
|
$ |
135,000,000 |
|
[**]
(2) For the avoidance of doubt, in the event that, for example, the first time Net Sales of the Product in the GSK Territory in any Calendar Year reaches €[**], is the same Calendar Year that Net Sales of the Product in the GSK Territory reaches €[**] for the first time, Sepracor shall be entitled to a milestone payment equal to $[**] for such Calendar Year (calculated by [**]).
(b) Reports and Payments. GSK shall notify Sepracor in writing within thirty (30) days after the achievement of each milestone set out in Section 7.2(a) by GSK, or any of its Affiliates or Sublicensees. GSK shall make payments within thirty (30) days of receipt of invoice issued by Sepracor following achievement of such milestone. Any milestone payable by GSK pursuant to Section 7.2(a) shall be made no more than once with respect to the achievement of each such milestone and no payment shall be due for a milestone which is not achieved. The milestone payments set forth in Section 7.2(a) shall not be refundable and shall not be creditable against future milestone payments, royalties or other payments to Sepracor under this Agreement.
(c) Target Label Dispute. Any dispute regarding the Target Label shall be resolved in the following manner:
(i) GSK and Sepracor shall each appoint a [**] (“Company RA Executives”) to meet to discuss whether the Target Label has been met. If, after [**], the Company RA Executives are unable to agree about whether the Target Label has been met, they shall agree upon one additional independent party, unaffiliated with either party, who is an acknowledged expert in product labeling within the EU (“Independent RA Expert”). If the Parties are unable to agree on the selection of an Independent RA Expert within a further [**],
CONFIDENTIAL
either Party may apply to the World Intellectual Property Organization Arbitration and Mediation Center to seek their appointment of an Independent RA Expert.
(ii) The Independent RA Expert shall meet with the Company RA Executives, alone, within [**] of his/her appointment and the group shall work for up to [**] to agree unanimously upon a conclusion. If the group is unable to make a unanimous decision in that period of time, then a majority vote shall prevail.
(iii) All costs of the above procedure to be borne by the party incurring them, except that the costs related to the Independent RA Expert are to be jointly and evenly shared.
7.3. Royalty Payments.
(a) Royalty Rate. Subject to the terms and conditions of this Agreement, in further consideration of the rights granted to GSK under this Agreement, GSK shall pay to Sepracor royalties at the rates set out below on Calendar Year Net Sales of the Product in the GSK Territory;
|
Calendar
Year Net Sales |
|
Royalty Rate |
|
|
On the first €[**] of Net Sales of Product in the GSK Territory in each Calendar Year |
|
[**] |
% |
|
On additional Net Sales of Product in the GSK Territory in each Calendar Year > €[**] but < €[**] |
|
[**] |
% |
|
On additional Net Sales of Product in the GSK Territory in each Calendar Year > €[**] |
|
[**] |
% |
(b) Royalty Term. GSK’s obligation to make royalty payments pursuant to Section 7.3(a) will commence upon the first commercial sale of the Product in the GSK Territory by GSK, its Affiliates or Sublicensees and will continue on a country-by-country basis for the Term.
(c) Royalty Rate Reduction. On a country-by-country basis, GSK’s royalty rate obligation set forth in Section 7(a) shall be reduced by [**]% upon the earlier occurring of:
(i) Upon the later of (I) the expiration of the last Valid Claim within a Patent Controlled by Sepracor that would be infringed by the licensed activities in such country
CONFIDENTIAL
in the absence of the licenses granted hereunder to GSK, or (II) ten (10) years post first launch in the GSK Territory.
(ii) Upon the total amount of Third Party sales of Generic Products in any country in the GSK Territory first exceeding [**] percent ([**]%) of the total sales by GSK, its Affiliates and Sublicensees of Product in such country in any Calendar Quarter, measured on a unit basis. For the purposes of this Section 7.3(c)(ii), the amount of sales of Products shall be ascertained by reputable published marketing data for such country (e.g. by reference to Standard Units data collected by IMS) or as otherwise mutually agreed. The term “Generic Product” refers to a pharmaceutical product, which is marketed by an entity other than GSK, its Affiliates or its Sublicensees, containing the Active Ingredient as the single therapeutically active ingredient, in a form and dosage capable of being prescribed as an alternative to the Product.
provided, however, such royalty reduction shall only be implemented once per country of the GSK Territory.
(iii) Net Sales in countries where a royalty rate reduction is applied pursuant to Section 7.3(c)(i) or 7.3(c)(ii) shall not be included in the calculation of Calendar Year Net Sales in GSK Territory for the purpose of determining royalty rate thresholds pursuant to Section 7.3(a).
(iv) A hypothetical example showing how royalties would be calculated in a situation where royalties are payable in full in some countries and at the reduced rate in others, is attached hereto as Schedule 7.3(c).
(d) Reports and Royalty Payment. During the Term of this Agreement, following the first commercial sale of the Product within the GSK Territory, within [**] after the end of each Calendar Quarter, GSK shall deliver to Sepracor a report setting out (on a country by country basis and in the aggregate) Net Sales in the relevant Calendar Quarter and all exchange rate conversions in accordance with Section 8.2(b). In addition, in such report GSK shall set out for [**]:
(i) units of the Product sold during the relevant Calendar Quarter;
(ii) gross sales figure of the Product in the relevant Calendar Quarter; and
(iii) a summary of deductions made in accordance with Sections 1.31 and 7.3(c).
All payment amounts due under Section 7.3(a) for such Calendar Quarter shall accompany such statement.
CONFIDENTIAL
(e) Limitation on Royalties; Annual Royalty Reconciliation.
(i) Subject to Section 7.3(e)(ii), in any given Calendar Year, the amount of the royalties paid hereunder plus the aggregate price paid by GSK for Bulk Tablets for Product sold in such Calendar Year shall not exceed [**]% of the aggregate Net Sales in such Calendar Year. On a dosage form basis, the aggregate price paid by GSK for Bulk Tablets for Product sold in such Calendar Year shall be determined by calculating the average price invoiced during such Calendar Year for the volume of Bulk Tablets required on average to make one unit of Product, and multiplying such average price by the number of units of Product sold during such Calendar Year. A hypothetical example showing how the above calculation would operate is attached hereto as Schedule 7.3(e).
(ii) Within [**] following the end of each Calendar Year, GSK shall provide Sepracor with a reconciliation of royalties due and royalties paid during such Calendar Year (the “Reconciliation Report”), including but not limited to the information required in Section 7.3(d) and a statement regarding (i) the aggregate price paid by GSK for Bulk Tablets for Product sold in such Calendar Year; (ii) the average price invoiced during such Calendar Year for the volume of Bulk Tablets required on average to make one unit of Product; and (iii) the number of units of Product sold during such Calendar Year. Copies of GSK’s reports setting forth its computation of such amounts shall be submitted to Sepracor with the Reconciliation Report. Unless Sepracor notifies GSK within [**] (the “Reconciliation Review Period”) after its receipt of the Reconciliation Report that it objects to the computation, the report shall be binding and conclusive.
(iii) During the Reconciliation Review Period, GSK shall permit an independent certified public accounting firm of internationally recognized standing selected by Sepracor and reasonably acceptable to GSK, at Sepracor’s expense, to have access to the relevant books and records of GSK (including, without limitation, records of barters and counter-trades) during GSK’s normal business hours to verify the Reconciliation Report. Sepracor will procure in such inspection to minimize disruption of the normal business activities of GSK to the extent reasonably practicable. The accounting firm shall disclose to Sepracor only whether the royalty reports are correct or incorrect and the specific details concerning any discreprencies. In the event that such independent accountant’s review of the Reconciliation Report determines that there has been an underpayment by GSK of royalties and that such underpayment exceeds five percent of the total amount owed for the Calendar Year, GSK shall reimburse Sepracor the reasonable and necessary fees and expensed of the independent accountant performing the audit.
(iv) If the reconciliation determines that payment is owed from one Party to the other, the Party owing payment shall pay the amount owed within thirty (30) days of the Parties’ agreement on the Reconciliation Report or the independent accountant’s determination of such amount.
7.4. Discounting. In the event that GSK or any of its Affiliates sell the Product to a Third Party as part of a bundle of products or services being purchased from GSK or its Affiliates, and GSK or its Affiliates discount the sales price of the Product to a greater degree than GSK or its Affiliate discounts the price of their other products or services to such customer then, in such case, the Net Sales for the sale of the Product to such Third Party shall be deemed
CONFIDENTIAL
to equal the arm’s length price that Third Parties would generally pay for the Product alone when not purchasing any other product or service from GSK or its Affiliate.
7.5. Third Party Licenses
(a) If either Party deems it necessary or advisable to obtain a license, or other rights to any Patent which may be or have the potential of being a Blocking Patent(s), such Party shall notify the other Party in writing. If the other Party agrees, then the Parties shall discuss in good faith the procedure by which the Parties will seek to obtain such license or other rights. The costs paid to a Third Party (including royalties, upfront, initial, milestones and periodic payments) associated with obtaining such license or other rights in relation to the GSK Territory shall be shared equally by the Parties.
(b) If the other Party disagrees with the initial assessment that it is necessary or advisable to obtain such license or rights to any Patent referred to in Section 7.5(a), the Party desiring the license shall obtain an opinion of independent patent counsel approved by the other Party (such approval not to be unreasonably withheld or delayed) to review infringement, validity and enforceability of the Patent(s) and the conclusions contained in such opinion shall be binding on both Parties. In the event that in the opinion of such independent patent counsel the identified Patent(s) may reasonably be expected to be held in subsequent legal proceedings to be infringed, valid and enforceable by any one of the activities licensed hereunder to GSK, then the Parties shall immediately proceed in the manner provided in Section 7.5(a) as if both Parties had initially agreed to seek the license or other rights. The fees, costs and expenses of the above independent patent counsel incurred in connection with this Section 7.5(b) shall be [**].
7.6. Third Party Technology. If after the Effective Date Sepracor or any of its Affiliates acquires an assignment of, or license under, Third Party Technology (other than Patents licensed or otherwise acquired pursuant to Section 7.5) for use in connection with the research, development, commercialization or manufacture of the Product in or for the GSK Territory, then Sepracor will promptly provide GSK with written notice of such acquisition and any additional financial terms to which Sepracor would be subject if GSK were to exploit a license under such Third Party Technology. If GSK desires to obtain such license, the Parties will promptly amend this Agreement to reflect such additional financial terms and to provide that the applicable Patents, know-how, data, or filings will be included in the definition of Sepracor Technology under this Agreement. For the avoidance of doubt, if GSK does not desire to obtain such license, such Third Party Technology shall not be included in the definition of Sepracor Technology under this Agreement.
7.7. Compulsory licenses.
(a) In the event that a governmental agency in any country in the GSK Territory grants or compels Sepracor to grant a compulsory license to any Third Party with respect to the Product, GSK shall have the benefit in such country of the terms granted to such Third Party to the extent that such terms are more favorable than those of this Agreement.
(b) If under applicable law in any country in the GSK Territory a governmental agency in any country grants or compels GSK to grant a compulsory license to any Third Party with respect to the Product, insofar as GSK is compensated on a royalty basis (as a percentage of the Sublicensee’s Net Sales) at a royalty rate lower than the royalty rate provided by Section 7.3, then the royalty rate to be paid to Sepracor by GSK on Net Sales in that country under Section 7.3 shall be reduced to the rate paid to GSK by the compulsory licensee.
7.8. Other Provisions. All royalties are subject to the following conditions:
(a) only one royalty shall be due with respect to the same unit of Product; and
(b) no royalties shall accrue on the disposition, in reasonable quantities, of Product by GSK, its Affiliate or Sublicensees as samples (promotion or otherwise) or as donations (for example, to non-profit institutions or government agencies for a non-commercial purpose) and without direct or indirect consideration.
ARTICLE VIII.
PAYMENTS, BOOKS AND RECORDS
8.1. Payment Method. All payments under this Agreement shall be made by bank wire transfer in immediately available funds to an account, capable of receiving United States Dollars, designated in writing by Sepracor. Payments hereunder will be considered to be made as of the day on which they are received by Sepracor’s designated bank.
8.2. Payment Currency: Currency Conversion.
(a) United States Dollars. Unless otherwise expressly stated in this Agreement, all amounts specified to be payable under this Agreement are in United States Dollars and shall be paid in United States Dollars.
(b) Currency Conversion. Net Sales in countries invoiced in currency other than United States Dollars or European Euros, as appropriate, shall be translated to United States Dollars or European Euros, as necessary, using [**] for the translation of foreign currency into United States Dollars or European Euros, as appropriate, as employed on a consistent basis by [**] and published accounts. GSK confirms that as of the Effective Date its current standard exchange rate methodology sources exchange rates from Reuters.
8.3. Fees, Expenses and Taxes
(a) Fees and Expenses. Each Party shall pay all fees and expenses incurred by such Party in connection with this Agreement, except as otherwise expressly provided herein.
CONFIDENTIAL
(b) Withholding Taxes. If laws or regulations require withholding by GSK of any taxes imposed upon Sepracor on account of any royalties or other payments paid under this Agreement, such taxes shall be deducted by GSK as required by law from such payment and shall be paid by GSK to the proper taxing authorities. Official receipts of payment of any withholding tax shall promptly be secured and promptly sent to Sepracor as evidence of such payment. The Parties will cooperate and exercise their reasonable efforts to ensure that any withholding taxes imposed are reduced as far as possible under the provisions of any applicable tax treaty and shall cooperate in filing any forms required for such reduction.
Sepracor warrants that Sepracor is resident for tax purposes in the United States and that Sepracor is entitled to relief from United Kingdom income tax under the terms of the double tax agreement between the United Kingdom and the United States. Sepracor shall notify GSK promptly in writing in the event that Sepracor ceases to be entitled to such relief.
Pending receipt of formal certification from the United Kingdom Inland Revenue, GSK may pay royalty income and any other payments under this Agreement to Sepracor by deducting tax at a rate specified in the double tax treaty between the United Kingdom and the United States. Sepracor agrees to indemnify and hold harmless GSK against any loss, damage, expense or liability arising in any way from a breach of the above warranties or any future claim by a United Kingdom tax authority or other similar body alleging that GSK was not entitled to deduct withholding tax on such payments at source at the treaty rate.
(c) Sales Taxes. All amounts to be paid under this Agreement are stated exclusive of any sales, use, value-added, goods and services or other applicable taxes, which shall be invoiced separately. Where appropriate, Sepracor shall provide GSK with a valid tax invoice. GSK shall upon receipt from Sepracor of a valid tax invoice pay to Sepracor the amount of any such applicable tax. Should any amounts of VAT paid by GSK to Sepracor be refunded subsequently by the fiscal authorities to Sepracor, Sepracor will refund these monies to GSK within thirty (30) days of receipt.
8.4. Records: GSK shall keep, and require its Affiliates and Sublicensees to keep, complete, true and accurate books of accounts and records for the purpose of determining the amounts payable to Sepracor pursuant to this Agreement. Such books and records shall be kept for such period of time required by law, but no less than at least [**] years following the end of the Calendar Quarter to which they pertain. Such records shall be subject to inspection in accordance with Section 7.3(e).
8.5. Late Payments. Any payments or portions thereof due under this Agreement that are not paid by the date such payment is due shall bear interest at an annual rate equal to the prime rate as published in the Wall Street Journal, Eastern edition, plus two percent (2%) per year, calculated on the number of days such payment is delinquent, compounded annually, and computed on the basis of a three hundred sixty five (365) day year. The provisions of this Section 8.5 shall in no way limit any other remedies available to Sepracor. The Parties agree that this provision, unless otherwise provided, will not apply to payments that are the result of a subsequent adjustment of an estimated payment, including rebates, adjustments, returns or
CONFIDENTIAL
reconciliations, nor to payments that are the subject matter of a good faith dispute between the Parties.
ARTICLE IX.
EXCLUSIVITY OBLIGATIONS
9.1 GSK Commitments.
(a) Subject to Section 9.1(b) below, on a country-by-country basis within the GSK Territory, during the Term of the Agreement, neither GSK nor any of its Affiliates shall promote, market or sell any Acquired Competing Product for any Product Indication.
(b) It shall not be a breach of Section 9.1(a) above for GSK or its Affiliates to promote, market or sell an Acquired Competing Product to which rights were acquired as part of an Acquisition Transaction until such time as GSK or its Affiliate has divested such Acquired Competing Product in accordance with Section 9.1(c) below.
(c) On a country-by-country basis within the GSK Territory, during the Term of the Agreement, in the event that GSK or its Affiliates acquire rights as part of an Acquisition Transaction to develop, promote, market or sell in a country in the GSK Territory any Acquired Competing Product, GSK or its Affiliates shall give written notice to Sepracor within [**] and use its reasonable endeavors to divest its rights to such Acquired Competing Product in such country within [**] of acquiring the rights to such Acquired Competing Product. If GSK or its Affiliates fail to divest such Acquired Competing Product within [**] of acquiring the rights to such Acquired Competing Product, and GSK or its Affiliates are actively promoting the Acquired Competing Product, Sepracor shall have the right to give ninety (90) days written notice to GSK terminating this Agreement on a country-by-country basis where the rights to such Acquired Competing Product have not been divested, provided that such termination shall not take effect in the event that during such notice period GSK or its Affiliates have divested the rights to such Acquired Competing Product.
(d) On a country by country basis within the GSK Territory, during the Term of this Agreement, neither GSK nor any of its Affiliates shall promote, market or sell, or enter into any agreement to promote, market or sell for the treatment of any Product Indication(s) any compound, other than the Product, which specifically and selectively targets [**].
(e) Subject to the above, GSK and its Affiliates retain the absolute freedom without restriction or limitation to research, develop, manufacture and commercialize products for Product Indication(s) which may compete with the Product in the GSK Territory or any part thereof (“GSK Competing Products”). In the event, however, that in any country in the GSK Territory during the Term GSK or any of its Affiliates file a MAA in which approval is sought for the marketing of a GSK Competing Product for a Product Indication, within [**] of filing such MAA, GSK shall notify the JSC whether GSK has decided to exercise its right to either: (i) terminate this Agreement in its entirety by providing twelve months’ notice to Sepracor; or (ii) to
CONFIDENTIAL
continue the commercialization of the Product, based on GSK’s medical and commercial assessment that the Product and the GSK Competing Product can adequately be positioned in the marketplace (“Commercialization Option”). In the event that the Commercialization Option is selected, GSK shall on a country-by-country basis make minimum royalty payments under Section 7.3(a) for each subsequent Calendar Year, or for part thereof, where GSK is simultaneously commercializing in such country the Product and a GSK Competing Product for one or more Product Indications, as stated below:
(I) During the period from the date of [**] until [**], the minimum annual royalty payment payable by GSK to Sepracor in relation to [**] would be [**] percent ([**]%) of the royalties received by Sepracor from GSK in respect of [**] during the full Calendar Year [**] in the [**].
(II) During the period from [**] until [**], the minimum annual royalty payment payable by GSK to Sepracor in relation to [**] would be [**] percent ([**]%) of the royalties received by Sepracor from GSK in respect of [**] during the full Calendar Year [**] in the [**].
(III) During the period following [**] until [**], the minimum annual royalty payment payable by GSK to Sepracor in relation to [**] would be [**] percent ([**]%) of the royalties received by Sepracor from GSK in respect of [**] during the full Calendar Year [**] in the [**].
For the avoidance of doubt, the above minimum annual royalty payments will be prorated for any Calendar Year in which GSK is simultaneously commercializing in a country the Product and a GSK Competing Product for one or more Product Indications, for part but not the whole Calendar Year.
(f) Sections 9.1(a)-(e) shall cease to apply on a country-by-country basis on the occurrence of any of the following:
(i) Expiry or termination, for whatever reason, of GSK’s rights under this Agreement to sell the Product in such country;
(ii) Launch of a Generic Product in such country;
(iii) The promotion, marketing or sale by Sepracor, its Affiliates or its sublicensees of any product with the Active Ingredient used in combination with another active ingredient for any Product Indication(s);
(iv) Any Regulatory Authority or court of competent jurisdiction in such country issuing a directive, order or request that the Product be withdrawn;
(v) GSK electing not to launch the Product pursuant to Section 6.4.
9.2. Sepracor’s Commitments. During the Term of the Agreement, neither Sepracor nor any of its Affiliates shall promote, market or sell, or enter into any agreement to promote, market or sell, in any country of the GSK Territory where the Agreement has not been terminated, any product containing the Active Ingredient as the sole therapeutic ingredient. However, this restriction shall not apply to any Excluded Products. For the avoidance of doubt, and consistent with the definition of Excluded Products, this provision is not intended to permit the promotion, marketing or sale of a product in which the Active Ingredient is simply co-packaged and/or co-administered with another product containing a different therapeutically active substance.
9.3. Non-prescription products. Sections 9.1 shall not apply in relation to products for promotion, marketing or sale without the legal requirement for a prescription.
ARTICLE X.
MANUFACTURE AND SUPPLY
10.1. General. During the Term of this Agreement, Sepracor shall at GSK’s request, and in accordance with the Supply Agreement, supply all of GSK’s, its Affiliates’ and its Sub-licensees’ requirements of Bulk Tablets, sufficient to enable GSK, its Affiliates and its Sub-licensees to meet their respective needs for Product for the GSK Territory.
10.2. Supply Agreement and Quality Agreement. Promptly following the Effective Date and within ninety (90) days thereof (or as otherwise agreed in writing by the Parties), GSK and Sepracor (or their respective Affiliates) shall negotiate in good faith and shall enter into a Supply Agreement (the “Supply Agreement”) and a Quality Agreement (the “Quality Agreement”) for the supply of Bulk Tablets by (or on behalf of) Sepracor for use in connection with Developmental Studies and for commercialization (including physician samples) of the Product in the GSK Territory, which shall reflect, among other things, the terms outlined in Schedule 10.2.
10.3. Supply Price.
(a) Bulk Tablets for use in Clinical Supplies, physician samples, or to be provided in support of Investigator Sponsored Clinical Studies shall be provided at [**]. Placebo for use in Clinical Supplies or to be provided in support of Investigator Sponsored Clinical Studies shall also be provided at [**].
(b) All other supplies of Bulk Tablets shall be provided at [**].
(c) Appropriate terms will be included in the Supply Agreement for calculation of [**] on an annual basis, using open book pricing.
CONFIDENTIAL
10.4. Minimum Price. Subject to Section 10.3, notwithstanding any provisions to the contrary in this Agreement, Sepracor shall not be required to supply Bulk Tablets to GSK at a cost below [**].
10.5. Pre-Supply Approval. GSK will have the right to conduct pre-supply approval audits of those manufacturing facilities of Sepracor, and any appointed third party manufacturer(s), which are used in the manufacture of Bulk Tablets for GSK, prior to taking supply. Thereafter, Sepracor shall not appoint any other third party manufacturer nor change the location of any part of the manufacturing process for the manufacture of Bulk Tablets for GSK without GSK’s prior written consent, not to be unreasonably withheld, and subject to the change control procedure set out in the Quality Agreement.
ARTICLE
XI.
CONFIDENTIALITY
11.1. Confidential Information. Except to the extent expressly authorized by this Agreement or otherwise agreed in writing, the Parties agree that the receiving Party (the “Receiving Party”) shall keep confidential and shall not publish or otherwise disclose or use for any purpose other than as provided for in this Agreement any confidential or proprietary information and materials, patentable or otherwise, in any form (written, oral, photographic, electronic, magnetic, or otherwise) which is disclosed to it by the other Party (the “Disclosing Party”) including, but not limited to, all information concerning the Product, information disclosed by one Party to the other pursuant to the Confidentiality Agreement, the contents of this Agreement and any other technical or business information of whatever nature (collectively, “Confidential Information”).
11.2. Development Data. Except to the extent expressly authorized by this Agreement or otherwise agreed in writing, the Parties agree that they shall keep confidential and shall not publish or otherwise disclose or use for any purpose other than as provided for in this Agreement any Data, results, reports or other know-how directly related to the Product which is generated in the performance of the Developmental Studies (“Development Data”).
11.3. Exceptions. Notwithstanding Sections 11.1 and 11.2, the obligations of confidentiality shall not apply to Confidential Information or Development Data that, in each case as demonstrated by competent evidence:
(a) was already known to the Receiving Party or any Affiliate, other than under an obligation of confidentiality, at the time of disclosure;
(b) was generally available to the public or was otherwise part of the public domain at the time of its disclosure to the Receiving Party;
CONFIDENTIAL
(c) became generally available to the public or otherwise part of the public domain after its disclosure by the Disclosing Party and other than through any act or omission of the Receiving Party or any Affiliate in breach of this Agreement;
(d) was subsequently lawfully disclosed to the Receiving Party or any Affiliate by a Person other than the Disclosing Party, and who, to the best knowledge of the Receiving Party, did not directly or indirectly receive such information from the Disclosing Party under an obligation of confidence; or
(e) is developed by the Receiving Party or its Affiliate without use of or reference to any information or materials disclosed by the Disclosing Party.
11.4. Permitted Disclosures. Notwithstanding the provisions of Section 11.1 above, and subject to Sections 11.5 and 11.6 below, each Party hereto may disclose Confidential Information of the other Party and Development Data to its Affiliates, permitted Sublicensees and any other Third Parties to the extent such disclosure: (a) is for the purpose of exercising the rights granted to it under this Agreement; (b) is reasonably necessary to prosecute or defend litigation; (c) is reasonably necessary to comply with applicable governmental laws or regulations; (d) is reasonably necessary to submit information to tax or other governmental authorities; or (e) is reasonably necessary to conduct clinical trials hereunder with respect to the Product. If a Party is required by law or regulations to make any such disclosure of Confidential Information of the other Party or Development Data, to the extent it may legally do so, it will give reasonable advance notice to the other Party of such disclosure and, save to the extent inappropriate in the case of patent applications or otherwise, will use its good faith efforts to secure confidential treatment of such Confidential Information or Development Data prior to its disclosure (whether through protective orders or otherwise) and disclose only the minimum amount of Confidential Information or Development Data necessary to comply with the requirement. For all other disclosures of Confidential Information of the other Party or Development Data permitted pursuant to this Section 11.4, including to Affiliates, permitted Sublicensees and other Third Parties, a Party shall ensure that the recipient thereof is bound by a written confidentiality agreement as materially protective of such Confidential Information or Development Data as this Article XI.
11.5. Confidentiality of this Agreement and its Terms. Except as otherwise provided in Section 11.6, each Party agrees not to disclose to any Third Party the existence of this Agreement or the terms of this Agreement without the prior written consent of the other Party hereto, except that each Party may disclose the terms of this Agreement: (a) to advisors (including financial advisors, attorneys and accountants) in each case under confidentiality obligations substantially equivalent to those in this Agreement; or (b) actual or potential acquisition or merger partners or Sublicensees on a need to know basis, in each case under written confidentiality agreements substantially equivalent to those in this Agreement.
CONFIDENTIAL
11.6. Public Announcements.
(a) As soon as practicable following the date hereof, the Parties will issue a joint press release announcing the existence of this Agreement. Except as required by law (including, without limitation, disclosure requirements of the U.S. Securities and Exchange Commission (“SEC”), the London, New York and NASDAQ stock exchanges, or any other stock exchange on which securities issued by a Party or its Affiliates are traded), neither Party shall make any other public announcement concerning this Agreement or the subject matter hereof without the prior written consent of the other, which shall not be unreasonably withheld or delayed; provided, that it shall not be unreasonable for a Party to withhold consent with respect to any public announcement containing any financial terms or any of such Party’s Confidential Information. In the event of a required public announcement, to the extent practicable under the circumstances, the Party making such announcement shall provide the other Party with a copy of the proposed text of such announcement sufficiently in advance of the scheduled release to afford such other Party a reasonable opportunity to review and comment upon the proposed text.
(b) The Parties will coordinate in advance with each other in connection with the redaction of certain provisions of this Agreement with respect to any filings with the SEC, the London, New York, and NASDAQ stock exchanges or any other stock exchange or governmental agency on which securities issued by a Party or its Affiliate are traded, and each Party will use reasonable efforts to seek confidential treatment for such terms; provided, that each Party will ultimately retain control over what information to disclose to the SEC, London, New York, and the NASDAQ stock exchange or any other stock exchange or governmental agency, as the case may be, and provided further that the Parties will use their reasonable efforts to file redacted versions with any governing bodies which are consistent with redacted versions previously filed with any other governing bodies. Other than such obligation, neither Party (or its Affiliates) will be obligated to consult with or obtain approval from the other Party with respect to any filings to the SEC, the NASDAQ stock exchange or any other stock exchange or governmental agency.
11.7. Publication of the Product Information. At least thirty (30) days prior to publishing, publicly presenting, and/or submitting for written or oral publication a manuscript, abstract or the like that includes Development Data or other information relating to the Product supplied or generated by GSK under this Agreement that has not been previously published, GSK shall provide to Sepracor a draft copy thereof for its review and approval (unless GSK is required by law to publish such information sooner). In addition, GSK shall, at the request of Sepracor, remove therefrom any Confidential Information of Sepracor (other than Development Data), except GSK shall have the right to publicly disclose any information, including Confidential Information, pertaining to safety of the Product that GSK has been advised by its legal counsel to disclose. For the avoidance of doubt, and without limiting in any way the above, GSK may post summaries on the GSK Clinical Trial Register of the results of any clinical trials conducted with respect to the Product pursuant to this Agreement without further approval from Sepracor.
11.8. Prior Non-Disclosure Agreements. As of the Effective Date of this Agreement, the terms of this Article XI shall supersede any prior non-disclosure, secrecy or confidentiality agreement between the Parties (or their Affiliates) dealing with the subject of this
CONFIDENTIAL
Agreement, including without limitation the Confidentiality Agreement. Any information disclosed under such prior agreements shall be deemed disclosed under this Agreement.
ARTICLE
XII.
PATENT PROSECUTION AND ENFORCEMENT
12.1. Ownership of Intellectual Property.
(a) Subject to the licenses granted to GSK pursuant to Section 2, Sepracor has, and shall retain all right, title and interest in and to, the Sepracor Technology (including without limitation the Sepracor Patents and Sepracor Know-How).
(b) Subject to Section 12.1(c) below, each Party shall have and retain all right, title and interest in all inventions, discoveries and know-how which are made, conceived, reduced to practice or generated by its employees, agents or other persons acting under its authority in the course of or as a result of this Agreement.
(c) Sepracor shall solely own all right, title and interest in and to any Non-Severable Improvements. GSK shall promptly disclose to Sepracor any and all Non-Severable Improvements conceived or reduced to practice by GSK, its Affiliates and Sublicensees, or any of its or their officers, directors, employees, consultants or other personnel in the performance of the Developmental Studies. All Non-Severable Improvements shall be the exclusive property of Sepracor and GSK shall take, and shall cause its Affiliates and Sublicensees to take, appropriate steps to ensure that its, or their officers, directors, employees, consultants and all other personnel are obligated to assign to Sepracor all right, title and interest each may have in any such Non-Severable Improvement and will cooperate to effect the foregoing. In consideration of this assignment, Sepracor agrees that in the event that the last Valid Claim within a Patent Controlled by Sepracor that would be infringed by the licensed activities in such country in the absence of the licenses granted under this Agreement covers a Non-Severable Improvement, then for the purposes of Section 7.3(c)(i), it shall be deemed that such Valid Claim has expired.
(d) GSK hereby grants Sepracor a worldwide, non-exclusive, irrevocable and fully paid up license, with right to sub-license, to use in relation to Sepracor’s, its Affiliate’s and sublicensees’ commercialization of the Product in the Sepracor Territory any Severable Improvements or Development Data which are made, conceived, reduced to practice or generated by GSK, its Affiliates and Sublicensees, or any of its or their officers, directors, employees, consultants or other personnel in the performance of the Development Studies.
(e) Joint Inventions. Subject to the rights granted under this Agreement, each Party can use, and grant non-exclusive licenses to use, an invention or discovery jointly owned by GSK and Sepracor pursuant to Section 12.1(b) without the other Party’s consent and has no duty to account to the other Party for such use or license, and each Party hereby waives any right it may have under the laws of any country to require any such consent or accounting.
CONFIDENTIAL
(f) Inventorship. For purposes of determining questions of inventorship for purposes of Section 12.1(b) the Parties shall apply the law of the country in which the patent applications directed to the relevant inventions are filed.
12.2. Prosecution and Maintenance of Sepracor Patents.
(a) Prosecution. Sepracor shall, [**], be responsible for the filing, prosecution and maintenance of all Sepracor Patents in the GSK Territory, [**]. GSK shall have the right to review new or pending patent applications in the GSK Territory and make recommendations to Sepracor concerning such patent applications. Sepracor will consider in good faith all reasonable suggestions of GSK with respect thereto, and agrees to keep GSK generally informed of the course of patent prosecution or other proceedings with respect to the Sepracor Patents within the GSK Territory, including without limitation providing confirmation that all renewal and maintenance fees have been paid in a timely fashion. GSK shall provide such patent consultation to Sepracor at no cost to Sepracor but shall have no obligation to provide such. GSK shall hold all information disclosed to it under this Section as Confidential Information. If Sepracor elects not to undertake the filing, prosecution and/or maintenance of any Sepracor Patents in the GSK Territory (or, after commencement of such filing, prosecution and/or maintenance, desires to cease the prosecution or the maintenance of any such Sepracor Patents in the GSK Territory), then Sepracor will notify GSK of such election with at least thirty (30) days’ prior written notice, identifying the specific Sepracor Patent(s) to which such election pertains and GSK will be, at its own expense and discretion, entitled to file, prosecute and/or maintain such Sepracor Patent(s) in the GSK Territory in the name of Sepracor, GSK, or both GSK and Sepracor, as the case may be.
(b) Patent Term Extensions. The Parties will discuss and recommend for which, if any, of the Patents within the Sepracor Patents in the GSK Territory should seek patent term extensions. Sepracor shall have the final decision-making authority whether, at its own expense, to file all applications and take actions necessary to obtain patent term extensions with respect to the Product pursuant to statutes in the GSK Territory, which extensions shall be owned by Sepracor. If Sepracor declines to pursue such patent term extensions in the GSK Territory, then GSK shall have the right at its own expense on behalf of Sepracor to file all such applications and take all such actions necessary to obtain such patent term extensions with respect to the Product. In each case, the Parties shall provide reasonable cooperation to the other Party which is seeking to obtain such extensions and additional protection. In the event that GSK has exercised the right to file, prosecute and/or maintain any Sepracor Patents pursuant to Section 12.2(a) above, the decision as to whether to pursue patent term extensions on such Sepracor Patents shall be at GSK’s sole discretion.
12.3. Patent Enforcement. In the event that Sepracor or GSK becomes aware of (i) an actual or threatened infringement or misappropriation of any Sepracor Technology in the GSK Territory by the manufacture, sale, offer for sale or use of any product (“Infringing Product”), or (ii) an actual or threatened challenge to any Sepracor Patents within the GSK Territory, that Party shall promptly notify the other Party in writing. Sepracor shall have the first right, but not the obligation, to initiate proceedings or take other appropriate action, at its own
CONFIDENTIAL
expense, against any Third Party taking or threatening to take such action. If Sepracor does not initiate proceedings or take other appropriate action within [**] of a request by GSK to initiate an enforcement proceeding or defense to a challenge to the Sepracor Patents then GSK shall be entitled, without obligation, to initiate infringement proceedings or take other appropriate action against an Infringing Product or Sepracor Patent challenge at its own expense and to include Sepracor as a nominal party plaintiff. The Party conducting such action shall have full control over its conduct, including settlement thereof; provided, however, that the Party conducting such action may not settle any such action, or make any admissions or assert any position in such action, in a manner that would materially adversely affect the rights or interests of the other Party whether within or outside the jurisdiction in which the action is brought, without the prior written consent of the other Party, which consent shall not be unreasonably withheld or delayed. In any event, the Parties shall assist one another and cooperate in any such litigation at the reasonable request of the other Party.
12.4. Trademark Enforcement and Protection. In the event that Sepracor or GSK becomes aware of (i) actual or threatened infringement, passing off, misappropriation or unfair advantage being taken of a Product Trademark or Other Trademark or misappropriation of any Product Trademark or Other Trademark in the GSK Territory by the manufacture, marketing, promotion, distribution, sale or use of any product (“Trademark Infringing Product”) or trademark (“Infringing Trademark”), or (ii) an actual or threatened challenge, such as an invalidity or revocation action, to any Product Trademark or Other Trademark within the GSK Territory, that Party shall promptly notify the other Party in writing. GSK shall have the first right, but not the obligation, to initiate proceedings or take other appropriate action, at its own expense, against any Third Party taking or threatening to take such action. Sepracor shall also have the right, but not the obligation, to participate in any legal proceedings in connection with such action initiated by GSK at Sepracor’s own expense and to make recommendations to GSK concerning such action; GSK shall consider in good faith all reasonable suggestions of Sepracor with respect thereto. If the applicable law in a country of the GSK Territory does not permit GSK, as an exclusive licensee, to initiate such actions, GSK shall be entitled to require Sepracor to initiate and diligently pursue such action on GSK’s behalf and at GSK’s expense. If GSK does not initiate proceedings or take other appropriate action within [**] of a request by Sepracor to initiate an enforcement proceeding or defense to a trademark challenge, then Sepracor shall be entitled, without obligation, to initiate infringement proceedings or take other appropriate action against a Trademark Infringing Product, Infringing Trademark or trademark challenge at its own expense and to include GSK as a nominal party plaintiff. The Party conducting such action shall have full control over its conduct, including settlement thereof; provided, however, that the Party conducting such action may not settle any such action, or make any admissions or assert any position in such action, in a manner that would materially adversely affect the rights or interests of the other Party whether within or outside the jurisdiction in which the action is brought, without the prior written consent of the other Party, which consent shall not be unreasonably withheld or delayed. In any event, the Parties shall assist one another and cooperate in any such litigation at the reasonable request of the other Party.
CONFIDENTIAL
12.5. Recovery. With respect to any suit or action referred to in Sections 12.3 and 12.4 any recovery obtained as a result of any such proceedings, by settlement or otherwise, shall be applied in the following order of priority:
(a) First, [**];
(b) Second, GSK shall recover [**], and Sepracor shall recover [**]; and
(c) Third, [**].
12.6. Cooperation. The Parties shall keep one another informed of the status of their respective activities regarding any litigation or settlement thereof concerning Sepracor Technology, Product Trademarks and Other Trademarks within the GSK Territory and shall assist one another and cooperate in any such litigation at the reasonable request of the other (including, without limitation, joining as a party plaintiff to the extent necessary and requested in writing by the other Party).
12.7. Infringement of Third Party Intellectual Property. In the event any action is taken by a Third Party against GSK or any Affiliate on the basis of alleged infringement of a Patent of such Third Party or of misappropriation of the know-how of a Third Party, or alleged infringement or passing off or misappropriation of a Third Party trademark, pursuant to the exercise by GSK or any Affiliate or Sublicensee of its rights hereunder, GSK shall promptly inform Sepracor and GSK shall be entitled at GSK’s expense to take whatever reasonable action it considers necessary or useful and Sepracor will provide any and all reasonable assistance, free of charge, to GSK. Sepracor shall also have the right, but not the obligation, to participate in any legal proceedings in connection with such action at its own expense and make recommendations to GSK concerning such action. GSK will consider in good faith all reasonable suggestions of Sepracor with respect thereto, and agrees to keep Sepracor informed of the course such action or legal proceedings with respect thereto. GSK shall control the legal proceedings, including the right to enter into any settlement, in order to be able to resume the exercise of the rights granted to GSK under the terms if this Agreement; provided, however, that GSK may not settle any such action, or make any admissions or assert any position in such action, in a manner that would adversely affect the rights or interests of Sepracor (whether within the GSK Territory or otherwise), or make any financial payment without the prior written consent of Sepracor, which consent shall not be unreasonably withheld or delayed. If as a result of such action or settlement, GSK has to pay any amount to the Third Party, including damages, license fees or royalties on sales of the Product, any and all such amounts as well as the cost related to such action or settlement shall be borne equally by Sepracor and GSK.
12.8. Patent Marking. GSK agrees to xxxx, and have its Affiliates and Sublicensees xxxx, all patented Products they sell or distribute pursuant to this Agreement in accordance with the applicable patent statutes and regulations in the country or countries of sale thereof.
CONFIDENTIAL
ARTICLE XIII.
TRADEMARKS
13.1. Cooperation. Through the JSC, the Parties will jointly determine whether the Product in each of the countries of the GSK Territory will use the Product Trademarks listed in Schedule 1.39, or other Product-specific trademarks (“Other Trademarks”). Upon the determination of an Other Trademark, or at the request of GSK any time thereafter, the Parties shall promptly amend this Agreement to update Part B of Schedule 1.39, at which time the representations and warranties provided by Sepracor under Section 16.2 shall be repeated in respect of such Other Trademarks.
13.2. Ownership. Sepracor shall own, and is hereby assigned, all right, title and interest in and to all trademarks used for the Product in the GSK Territory, including, the Product Trademarks and Other Trademarks, and including any intellectual property rights in the Product Trademarks and Other Trademarks, but not including any company marks or logos of GSK or its Affiliates.
13.3. Registration of Trademarks of the Product in the GSK Territory. Sepracor shall, in a timely manner, file, seek registration and maintain, for the Term of this Agreement and for any time GSK continues to use the Product Trademarks and Other Trademarks in accordance with the Wind-down Period in Section 15.2(a) and/or Section 15.3(a), [**], appropriate registrations and prosecute and defend oppositions to applications for such registrations for the Product Trademarks and Other Trademarks in the GSK Territory. For the avoidance of doubt, Sepracor shall perform such actions in all countries in the GSK Territory where the Product Trademarks and Other Trademarks will be used in accordance with this Agreement and shall file applications in time for relevant XXXx to be granted and for launch of the Product to progress in accordance with the Commercialization Plan. At GSK’s request, Sepracor will keep GSK informed of the status of such applications, registrations, actions and relevant proceedings.
13.4. Grant of License. Subject to the terms and conditions of this Agreement, Sepracor hereby grants to GSK an exclusive license to use the Product Trademarks and Other Trademarks, including as a domain name or part thereof, to package, market, promote, distribute, import, offer for sale and sell the Product in the GSK Territory during the Term and in accordance with this Agreement. Any use of the Product Trademarks and Other Trademarks shall inure to the sole benefit of Sepracor.
13.5. Trademark Restrictions. During the Term of this Agreement, Sepracor may not use, or authorize a Third Party not already authorized to use, the Product Trademarks and Other Trademarks (or any marks which are confusingly similar to the Product Trademarks and Other Trademarks taking into account the nature of the use, including the similarity or dissimilarity of the relevant goods) in any country of the GSK Territory or Sepracor Territory for the packaging, marketing, promotion, distribution, importation, offering for sale and sale of the Product or any other product. Sepracor also agrees not to use or authorize another to use during the Term of this Agreement, the LUNESTA® trademark (or any confusingly similar xxxx taking
CONFIDENTIAL
into account the nature of the use, including the similarity or dissimilarity of the relevant goods) in any country of the GSK Territory for the packaging, marketing, promotion, distribution, importation, offering for sale and sale of any products within the GSK Territory. For the avoidance of doubt, where this Agreement allows Sepracor or GSK to terminate the Agreement in a particular country, then upon such termination and in conjunction with the transfer obligations in Section 13.9 herein, Sepracor shall have the right to use or allow the use of any Product Trademark, Other Trademark or the LUNESTA® trademark in such country. Sepracor shall promptly notify GSK in the event that Sepracor becomes aware that any Third Party to which Sepracor has, as of the Effective Date, already authorized to use the Product Trademarks and/or Other Trademarks in the Sepracor Territory is using or planning to use the Product Trademarks and/or Other Trademarks for the packaging, marketing, promotion, distribution, importation, offering for sale and sale of the Product or any other product. In such event, Sepracor will cooperate in good faith and use its reasonable endeavours to resolve with GSK and the Third Party any concerns that GSK has regarding the use of the Product Trademarks and/or Other Trademarks by such Third Party.
13.6. Recordation of Licenses. If a trademark license must be recorded in the GSK Territory, or if GSK wishes to record its interest as a licensee on a trademarks register anywhere in the GSK Territory and a trademark licence is required to do so, Sepracor will provide reasonable assistance to allow GSK to become recorded as a licensee of the Product Trademarks and Other Trademarks on any trademarks register in the GSK Territory, including, without limitation, promptly providing to GSK, at GSK’s written request, a separate trademark license for the Product Trademarks and/or Other Trademarks on terms that are consistent with, and no broader or more onerous than, the terms of this Agreement, and GSK will arrange for the prompt recordation of such trademark license with the appropriate governmental agency, at GSK’s expense. GSK shall cooperate in the preparation and execution of such documents. No such recordation shall give GSK any ownership or claim to ownership of the Product Trademarks or Other Trademarks.
13.7. Display. All packaging materials, labels and promotional materials for the Product in the GSK Territory shall display the Product Trademarks and/or Other Trademarks approved by the JSC and no other product-specific trademarks or branding. In addition, such packaging materials, labels and promotional materials shall display the trade name(s) of GSK, or its Affiliates or Sublicensees, in reasonable size and prominence as allowed by applicable local regulations. The trademarks of GSK, trade dress, style of packaging and the like with respect to the Product in the GSK Territory may be determined by GSK in a manner that is consistent with GSK’s standard trade dress and style. All packaging materials, labels and promotional materials for the Product in the GSK Territory shall display the following: “Manufactured and sold under license from Sepracor Inc.” and “[the Product Trademark or Other Trademark] is a trademark of Sepracor Inc.”. GSK shall make no other use of Sepracor’s name or logo on packaging materials.
13.8. Approval of Packaging. So that Sepracor can properly evaluate the protection of its trademarks and other trade dress, GSK shall submit representative examples of packaging and the Product displaying the Product Trademarks and/or Other Trademarks, to
CONFIDENTIAL
Sepracor for review and written approval at least [**] prior to the first use of such packaging for or on the Product and [**] prior to use of any subsequent packaging which are a material change to such packaging. For the avoidance of doubt, such review does not extend to a right to review and approve the trademarks, trade dress and style of packaging and the like of GSK which may be determined by GSK in accordance with Section 13.7. Sepracor shall not unreasonably withhold or delay approval, but in any event, if Sepracor has not responded in writing within [**] after the submission to Sepracor of such packaging for or on the Product, Sepracor’s approval will be deemed to have been received.
13.9. Termination of Trademark License. GSK’s right to use the Product Trademarks and Other Trademarks) shall terminate on a country-by-country basis when GSK’s rights to the Product pursuant to this Agreement are terminated or expire in any country. GSK shall take all such steps as Sepracor may reasonably request to give effect to the termination of any license to the Product Trademarks and Other Trademarks, and to record any documents that may be required to evidence the termination of such licenses and transfer to Sepracor of all rights, registrations, recordation and the like for such Product Trademarks and Other Trademarks.
ARTICLE XIV.
TERM AND TERMINATION
14.1. Term. This Agreement shall commence on the Effective Date and, unless earlier terminated pursuant to Sections 14.2, 14.3 or 14.4, shall continue in each country of the GSK Territory on a country by country basis until the later of (a) the tenth anniversary of the Effective Date, and (b) the date on which GSK ceases to continue to market, promote, distribute, import or sell the Product in any country of the GSK Territory (the “Term”).
14.2. Early Termination. Each Party shall have the right to terminate this Agreement in its entirety before the end of the Term;
(a) by mutual written agreement of the Parties;
(b) upon written notice by either Party if the other Party is in material breach of this Agreement and has not cured such breach after notice from the terminating Party requesting cure of the breach; provided, however, in the event of a good faith dispute with respect to the existence of a material breach, the cure period shall be tolled until such time as the dispute is resolved pursuant to Section 18; and provided that the terminating Party has given the defaulting Party the following opportunities to remedy any breach:
(i) the written notice of breach referenced above shall detail the specific obligation under this Agreement which is alleged to have been breached; the manner of such alleged breach; and the steps which must be taken in order to remedy such breach; and
CONFIDENTIAL
(ii) the terminating Party has provided the defaulting Party with a reasonable amount of time (but no more than [**]) in which (x) to complete any steps which might be taken to remedy the breach, as stated in the notification of breach, or (y) if completion of those steps is not possible within a [**] period, to commence those steps required as stated in the notification of breach, on the condition that the defaulting Party continues to perform those steps with due diligence and the breach can be cured within a mutually agreeable period of time; or
(c) upon the bankruptcy or insolvency, or the filing of an action to commence insolvency proceedings against the other Party, or the making or seeking to make or arrange an assignment for the benefit of creditors of the other Party, or the initiation of proceedings in voluntary or involuntary bankruptcy, or the appointment of a receiver or trustee of such Party’s property that is not discharged within ninety (90) days.
14.3. Other GSK Termination Rights.
(a) GSK has the right to terminate this Agreement in its entirety at any time prior to the earlier of (i) first launch of the Product in the GSK Territory or (ii) the date six months after EMEA approval of the Product, by giving at least sixty (60) days’ written notice to Sepracor.
(b) On a country-by-country basis, GSK has the right to terminate this Agreement in respect of any country in the GSK Territory in which the Product has not been launched at the time of providing notice of termination, by giving at least sixty (60) days’ written notice to Sepracor.
(c) On a country-by-country basis, GSK has the right to terminate this Agreement in respect of any country in the GSK Territory in which the Product has already been launched at the time of providing notice of termination, by giving at least one hundred and eighty (180) days’ written notice to Sepracor. For the avoidance of doubt, in relation to any country in which the Commercialization Option has been exercised, if GSK exercises its right to terminate under this Section 14.3(c) during the twelve month period following GSK’s decision to exercise the Commercialization Option pursuant to Section 9.1(e)(ii), the termination under this Section 14.3(c) shall not take effect until the later of: (i) expiry of the (180) days’ written notice provided hereunder and (ii) expiry of the twelve month period following GSK’s decision to exercise the Commercialization Option pursuant to Section 9.1(e)(ii).
(d) GSK has the right to terminate this Agreement, on a country-by-country basis, at any time upon ten (10) days’ written notice to Sepracor, if the applicable Regulatory Authority decides to withdraw the Product from such country in the GSK Territory.
(e) GSK has the right to terminate this Agreement with immediate effect in its entirety, or on a country-by-country basis, upon termination of the Supply Agreement for any reason.
CONFIDENTIAL
(f) Pursuant to and on the terms of Section 9.1(e).
14.4. Other Sepracor Termination Rights.
(a) Sepracor may terminate this Agreement in its entirety upon thirty (30) days prior written notice to GSK, if GSK or its Affiliates challenge any of the Product Trademarks or Sepracor Technology that relates to the Product, including, without limitation, the Sepracor Patents specified on Schedule 1.44 of this Agreement. Any such termination shall only become effective if GSK and its Affiliates have not withdrawn such challenge before the end of the above notice period.
(b) Sepracor may terminate this Agreement in its entirety upon sixty (60) days prior written notice to GSK, if any of GSK’s Sublicensees challenge any of the Product Trademarks or Sepracor Technology that relates to the Product, including, without limitation, the Sepracor Patents specified on Schedule 1.44 of this Agreement. Any such termination shall only become effective if GSK’s Sublicensee has not withdrawn such challenge before the end of the above notice period.
(c) Sepracor may terminate this Agreement in its entirety upon sixty (60) days prior written notice to GSK, if launch of the Product has not occurred in [**] within the [**] period following EMEA approval. However, for the avoidance of doubt, failure to [**] shall not be counted under this Section 14.4(c) if GSK has given proper notice under Section 6.4(a) and complied with the terms of that Section in respect of that country.
(d) Pursuant to and on the terms of Section 4.3, Sepracor may terminate this Agreement in relation to the European Union in the event that GSK notifies Sepracor that it does not wish to undertake the Required Studies.
(e) Pursuant to and on the terms of Section 5.3, Sepracor may terminate this Agreement on a country-by-country basis in the event that GSK notifies Sepracor that it does not wish to undertake the Required Studies in such country.
(f) Pursuant to and on the terms of Section 6.4(b).
(g) Pursuant to and on the terms of Section 9.1(c).
ARTICLE XV.
EFFECT OF TERMINATION
15.1. Accrued Obligations. The termination of this Agreement, in whole or part, for any reason shall not release either Party from any liability which, at the time of such termination, has already accrued to such Party or which is attributable to a period prior to such termination, nor will any termination of this Agreement preclude either Party from pursuing all
CONFIDENTIAL
rights and remedies it may have under this Agreement, at law or in equity, with respect to breach of this Agreement.
15.2. Rights on Termination. This Section 15.2 shall apply upon the termination of GSK’s rights under this Agreement if, and only if, such termination is made by Sepracor pursuant to Sections 14.2(b), 14.4(a), 14.4(b), or 14.4(g) or is made by GSK pursuant to Section 14.3(c):
(a) Wind-down Periods.
(i) Development. In the event there are any on-going Developmental Studies of the Product in the GSK Territory (or, if the termination is limited to a specific country, within such country), at Sepracor’s request in writing, GSK agrees: (A) to complete, [**], any such Developmental Studies, or (B) to the extent so requested by Sepracor, to promptly transition, [**], to Sepracor or its designee such Developmental Studies or portions thereof for Sepracor or its designee to complete [**]. If GSK shall continue to conduct any such Developmental Studies, it shall do so in accordance with the terms and conditions of this Agreement; provided, however, that in no case shall GSK be obligated to pursue such activities for a period exceeding [**] after the date of notice of such termination.
(ii) Packaging. GSK shall, at the written request of Sepracor, continue to package the Bulk Tablets at costs to be negotiated in good faith at the time until Sepracor notifies GSK in writing that it has made other arrangements. GSK’s packaging obligations under this Section 15.2(a)(ii) shall not exceed [**] following the effective date of the termination. Upon notification by Sepracor or the expiry of the [**], whichever is sooner, GSK shall cease packaging of the Product unless the parties mutually agree terms under which packaging may be continued.
(iii) Commercialization. GSK, its Affiliates and Sublicensees, shall continue, to the extent that GSK, its Affiliates and Sublicensees continue to have stocks of usable Product, to fulfill orders received from customers for the Product in the GSK Territory (or, if the termination is limited to a specific country, within such country) until up to [**] days after the later of (A) the date on which Sepracor notifies GSK in writing that Sepracor has secured an alternative distributor or licensee for the Product in the GSK Territory and (B) GSK has initiated transition of the XXXx and Marketing Approvals for the Product in the GSK Territory (or, if the termination is limited to a specific country, within such country) to such distributor or licensee, but in no event for more for than [**] after the date of notice of termination. For the Products sold by GSK after the effective date of a termination (i.e., after the expiration of the applicable termination notice period), GSK shall continue to pay royalties on the amount of Net Sales from such sales pursuant to Section 7.3. Notwithstanding the foregoing, GSK, its Affiliates and its Sublicensees shall cease such activities in the GSK Territory (or, if termination is limited to a specific country, within such country), as the case may be, upon [**] written notice given by Sepracor at any time after the effective date of a termination requesting that such activities (or portion thereof) cease. In the case of a termination of this Agreement in its entirety, within [**] after Sepracor has given notice to GSK requesting the cessation of activities pursuant to the
CONFIDENTIAL
provision of this Section, GSK shall notify Sepracor of an estimate of the quantity of the Product and its shelf life remaining in GSK’s inventory and Sepracor shall have the right to purchase any such quantities of the Product from GSK for [**], being the amount [**]. To the extent Sepracor does not purchase such quantities, GSK may sell such quantities in the GSK Territory during the [**] after the effective date of such termination within the shelf life remaining for the Product.
(b) Assignment of Filings and Marketing Approvals. As soon as practicable following the effective date of termination, GSK shall assign or cause to be assigned to Sepracor or its designee (or to the extent not so assignable, GSK shall take all reasonable actions to make available to Sepracor or its designee the benefits of) all regulatory filings and registrations (including INDs, XXXx and Marketing Approvals) for the Product in the GSK Territory (or, if termination is limited to a specific country, within such country), including any such regulatory filings and registrations made or owned by its Affiliates and/or Sublicensees. Sepracor shall notify GSK before the effective date of termination, whether the regulatory filings and registrations should be assigned to Sepracor or its designee, and if the latter, identify the designee, and provide GSK with all necessary details to enable GSK to effect the assignment (or availability). If Sepracor fail to provide such notification prior to the effective date of termination, GSK shall assign the regulatory filings and registrations to Sepracor.
(c) Transition. GSK shall use diligent efforts to cooperate with Sepracor and/or its designee to effect a smooth and orderly transition in the development, sale and marketing, promotion and commercialization of the Product in the GSK Territory (or, if termination is limited to a specific country, within such country) during the notice and the Wind-down Periods. Without limiting the foregoing, GSK shall use diligent efforts to conduct, in an expeditious manner, any activities to be conducted under this Section 15.2. Sepracor shall use diligent efforts to identify and finalize an agreement or other arrangement with a Third Party in relation to the Product and/or, to the extent Sepracor is able to take over such activities under applicable law or regulations, take over, directly or through an Affiliate, all activities related to the Product, and in particular development activities on-going at the time of the effective date of the termination and the transfer of the regulatory filings and registrations (including INDs, XXXx and Marketing Approvals) into the name of Sepracor or Sepracor’s designee so that the Wind-down Period be as limited as possible.
(d) Rights Become Non-Exclusive. Notwithstanding any other provision of this Agreement, following the effective date of termination and during the Wind-down Periods, GSK’s, its Affiliates’ and Sublicensees’, rights with respect to the Product in the GSK Territory (or, if termination is limited to a specific country, within such country) shall be non-exclusive, and, without limiting the foregoing, Sepracor shall have the right to engage one or more other distributors and/or licensees of the Product in all or part of the GSK Territory.
(e) Continuing Payment Obligations. Any Product sold or disposed of by GSK, its Affiliates or Sublicensees, in the GSK Territory in accordance with this Section 15.2 shall be subject to the applicable payment obligations under Article VII above.
CONFIDENTIAL
(f) Licenses. Sepracor hereby grants to GSK a limited, non-exclusive license under the Sepracor Technology, the Product Trademarks and Other Trademarks, from the effective date of any termination until the expiration of the applicable Wind-down Period(s), solely for the purposes of permitting GSK to comply with its obligations under this Section 15.2 and Section 15.3. Effective as the date of transfer of any packaged Product from GSK to Sepracor pursuant to Section 15.2(a)(iii), GSK hereby grants to Sepracor a limited, non-exclusive license to use any trade names, trademarks, or other identifying markings of GSK or its Affiliates included in the packaging and labeling of such Product, solely for the purposes of selling and distributing any Product, within the shelf life remaining for the Product, which has already been packaged by GSK, its Affiliates or Sublicensees to include such markings as at the date of transfer; the “use” rights granted by GSK do not include a right to print any GSK trade names, trademarks or other identifying markings of GSK, its Affiliates or Sublicensees.
(g) Sublicensees; Third Party Agreements. Upon the completion of the obligations defined in Sections 15.2 and 15.3, any contracts with Sublicensees of the Product in the GSK Territory engaged by GSK shall terminate at the same time as the Agreement with GSK. At the written request of Sepracor, GSK will assign any Product-specific Third Party Agreements requested by Sepracor for assignment to the furthest extent possible, provided that such assignment is permitted under the Product-specific Third Party Agreement or accepted by the Third Party. In the event such assignment is not requested by Sepracor or is not accepted by such Third Party, then the rights of such Third Party with respect to the Product shall terminate upon termination of GSK’s rights. GSK shall ensure that its Affiliates, Sublicensees and such Third Party (if its contract is not assigned to Sepracor pursuant to this Section 15.2(g)) shall transition any remaining Product back to Sepracor in the manner set forth in this Section 15.2(a)(iii) as if such Affiliate or Third Party were named herein. GSK shall include provisions requiring compliance with these provisions in the agreements with Sublicensees and Third Parties.
15.3. Rights on Termination under Section 14.3(b). This Section 15.3 shall apply upon the termination of GSK’s rights under this Agreement if, and only if, such termination is made by GSK pursuant to Section 14.3(b):
(a) Wind-down Period for Development. In the event there are any on-going Developmental Studies of the Product in the GSK Territory (or, if the termination is limited to a specific country, within such country), at Sepracor’s request in writing, GSK agrees: (A) to complete, at GSK’s expense, any such Developmental Studies, or (B) to the extent so requested by Sepracor, to promptly transition, at GSK’s expense, to Sepracor or its designee such Developmental Studies or portions thereof for Sepracor or its designee to complete at their expense. If GSK shall continue to conduct any such Developmental Studies, it shall do so in accordance with the terms and conditions of this Agreement; provided, however, that in no case shall GSK be obligated to pursue such activities for a period exceeding twelve (12) calendar months after the date of notice of such termination.
15.4. No Renewal, Extension or Waiver. Acceptance of any order from, or sale or license of any the Product to GSK after the notice or effective date of termination of this
CONFIDENTIAL
Agreement shall not be construed as a renewal or extension hereof, or as a waiver of termination of this Agreement.
15.5. Survival. Upon the termination of this Agreement, all rights and obligations of the Parties under this Agreement shall terminate for the country(ies) effected by such termination, except those described in the following Articles and Sections: Articles I, VIII, XI, XIV, XV, XVI, XVII, XVIII, XIX and XX and Sections 5.14, 12.1 and 13.2. Termination shall not affect any rights to receive payments that have accrued under Sections 7.2 and 7.3 at the time of termination. The survival of rights and obligations under the Supply Agreement and Quality Agreement shall be determined under the terms of such agreements.
ARTICLE XVI.
REPRESENTATIONS, WARRANTIES AND COVENANTS
16.1. General Representations. Each Party hereby represents and warrants to the other Party, as of the Effective Date, as follows:
(a) Duly Organized. Such Party is a corporation duly organized, validly existing and in good standing under the laws of the jurisdiction of its incorporation, is qualified to do business and is in good standing as a foreign corporation in each jurisdiction in which the conduct of its business or the ownership of its properties requires such qualification and failure to have such would prevent such Party from performing its obligations under this Agreement.
(b) Due Execution Binding Agreement. This Agreement is a legal and valid obligation binding on such Party and enforceable in accordance with its terms. The execution, delivery and performance of this Agreement by such Party have been duly authorized by all necessary corporate action and do not and will not: (i) require any consent or approval of its stockholders; (ii) to such Party’s knowledge and belief, violate any law, rule, regulation, order, writ, judgment, decree, determination or award of any court, governmental body or administrative or other agency having jurisdiction over such Party; nor (iii) conflict with, or constitute a default under, any agreement, instrument or understanding, oral or written, to which such Party is a party or by which it is bound.
(c) Consents. Such Party has obtained, or is not required to obtain, the consent, approval, order or authorization of any Third Party, or has completed, or is not required to complete any registration, qualification, designation, declaration, or filing with, any Regulatory Authority or governmental authority, in connection with the execution and delivery of this Agreement and the performance by such Party of its obligations under this Agreement.
(d) Debarment. Such Party is not debarred under the United States Federal Food, Drug and Cosmetic Act and it does not, and will not during the term of this Agreement, employ or use the services of any Person who is debarred, in connection with the development, manufacture or commercialization of the Product. In the event that either Party becomes aware of the debarment or threatened debarment of any Person providing services to such Party,
CONFIDENTIAL
including the Party itself and its Affiliates or Sublicensees, which directly or indirectly relate to activities under this Agreement, the other Party shall be immediately notified in writing.
16.2. Representations and Warranties of Sepracor. Sepracor represents and warrants to GSK that, as of the Effective Date:
(a) it has the full right and authority to grant the rights and licenses provided herein;
(b) Sepracor is the exclusive owner of all right, title and interest in the Sepracor Patents listed in Schedule 1.44. To the best of Sepracor’s knowledge, the issued claims in the Sepracor Patents listed in Schedule 1.44 are valid and enforceable, and the patent applications have been duly filed;
(c) to the best of Sepracor’s knowledge, neither the development, use, sale or import of the Product in the GSK Territory, as currently being manufactured and commercialized in the Sepracor Territory, infringes any Third Party’s valid patents or constitutes a misappropriation of a Third Party’s trade secrets or other intellectual property rights. For purposes of this Section 16.2(c) and Section 16.2(d) below, the phrase “to the best of Sepracor’s knowledge” means the actual knowledge of the attorneys in Sepracor’s legal department and the members of Sepracor’s corporate senior management team;
(d) to the best of Sepracor’s knowledge, in the GSK Territory, there is no Third Party infringing any of the Sepracor Patents or misappropriating Sepracor Know-How in derogation of the rights granted to GSK in this Agreement with respect to the Product;
(e) Sepracor is the exclusive owner of all right, title and interest in the Product Trademarks. The Product Trademarks are available for valid registration, or have been validly registered, by Sepracor in all the Major Markets except as set forth on Schedule 16.2(g);
(f) it has not previously granted any right, license or interest in or to the Sepracor Technology, or any portion thereof, or the Product Trademarks, or any confusing similar trademarks, that is in conflict with the rights or licenses granted to GSK under this Agreement;
(g) except as set forth on Schedule 16.2(g), there are no actual or to the best of Sepracor’s knowledge, threatened, anticipated, pending or alleged actions, suits, claims, interference proceedings or governmental investigations in the GSK Territory involving the Product, Sepracor Technology or the Product Trademarks by or against Sepracor, or any of its Affiliates;
(h) to Sepracor’s knowledge, there is no actual, pending, alleged or threatened product liability action nor intellectual property right litigation in relation to the Product, all the foregoing being applicable for the GSK Territory;
CONFIDENTIAL
(i) to Sepracor’s knowledge, there is no actual, pending, alleged or threatened infringement in the GSK Territory by a Third Party of any of the Sepracor Technology or Product Trademarks relating to the Product in the GSK Territory; and
(j) Sepracor has not failed to furnish GSK with any information requested by GSK, or intentionally concealed from GSK, any information in its possession which Sepracor reasonably believes would be material to GSK’s decision to enter into this Agreement and undertake the commitments and obligations set forth herein
16.3. Representations and Warranties of GSK. GSK represents and warrants to Sepracor that, as of the Effective Date it has the full right and authority to grant the rights granted herein.
16.4. DISCLAIMER. EXCEPT AS EXPRESSLY SET FORTH IN THIS AGREEMENT, OR ANY OTHER AGREEMENT CONTEMPLATED HEREUNDER, NEITHER PARTY MAKES ANY REPRESENTATIONS OR EXTENDS ANY WARRANTIES OF ANY KIND, EITHER EXPRESS OR IMPLIED, AND EACH PARTY EXPRESSLY DISCLAIMS ALL IMPLIED WARRANTIES OF MERCHANTABILITY AND OF FITNESS FOR A PARTICULAR PURPOSE OR USE, NON-INFRINGEMENT, VALIDITY AND ENFORCEABILITY OF PATENTS, OR THE PROSPECTS OR LIKELIHOOD OF DEVELOPMENT OR COMMERCIAL SUCCESS OF THE PRODUCT. IN NO EVENT WILL EITHER PARTY BE LIABLE FOR THE OTHER PARTY’S LOST PROFITS, LOSS OF DATA, OR FOR ANY SPECIAL, INDIRECT, INCIDENTAL, CONSEQUENTIAL OR PUNITIVE DAMAGES, HOWEVER CAUSED, ON ANY THEORY OF LIABILITY AND WHETHER OR NOT SUCH PARTY HAS BEEN ADVISED OF THE POSSIBILITY OF SUCH DAMAGES, ARISING UNDER ANY CAUSE OF ACTION AND ARISING IN ANY WAY OUT OF THIS AGREEMENT. THE FOREGOING LIMITATIONS WILL NOT LIMIT EITHER PARTY’S OBLIGATIONS TO THE OTHER PARTY UNDER SECTION XVII OF THIS AGREEMENT.
16.5. Prior Knowledge. No liability for any breach of any representation or warranty given by a Party under this Section 16 shall arise to the extent to which the other Party has knowledge at the Effective Date that such representation or warranty is untrue or inaccurate.
ARTICLE XVII.
INDEMNIFICATION; RECALLS
17.1. Indemnification of Sepracor. GSK shall indemnify and hold harmless each of Sepracor, its Affiliates and the directors, officers, shareholders and employees of such entities and the successors and assigns of any of the foregoing (the “Sepracor Indemnitees”), from and against any and all liabilities, damages, penalties, fines, costs and expenses (including, reasonable attorneys’ fees and other expenses of litigation) (“Liabilities”) from any claims,
CONFIDENTIAL
actions, suits or proceedings brought by a Third Party (a “Third Party Claim”) incurred by any Sepracor Indemnitee, arising from, or occurring as a result of: (a) packaging, use, marketing, distribution, importation, offer for sale or sale of the Product by GSK, its Affiliates or Sublicensees including, any Products Liability Claim (subject to and shared in accordance with the mechanism set forth in Section 17.5, below); (b) the performance of any Developmental Study regarding the Product commenced by GSK, its Affiliates or Sublicensees and (c) any material breach of any representations, warranties or covenants by GSK under this Agreement; except to the extent such Third Party Claims fall within the scope of the indemnification obligations of Sepracor set forth in Section 17.2, below, or result from the gross negligence or willful misconduct of a Sepracor Indemnitee.
17.2. Indemnification of GSK. Sepracor shall indemnify and hold harmless each of GSK, its Affiliates and Sublicensees, and the directors, officers and employees of GSK, its Affiliates and Sublicensees, and the successors and assigns of any of the foregoing (the “GSK Indemnitees”), from and against any and all Liabilities from any Third Party Claims incurred by any GSK Indemnitee, arising from, or occurring as a result of: (a) the manufacturing, use, marketing, distribution, offer for sale or sale of the Product by Sepracor, an Affiliate or its licensee, including any Products Liability Claim (subject to and shared in accordance with the mechanism set forth in Section 17.5, below); (b) the development of the Product by Sepracor (but only where not performed under GSK’s direction); (c) the use of the Product Trademarks by GSK in the GSK Territory in accordance with this Agreement, and (d) any material breach of any representations, warranties or covenants by Sepracor under this Agreement, except to the extent such Third Party Claims falls within the scope of the indemnification obligations of GSK set forth in Section 17.1, above, or result from the gross negligence or willful misconduct of an GSK Indemnitee.
17.3. Procedure. Except with respect to the Product Liability Claims subject to Section 17.5, below, a Party that intends to claim indemnification under this Article XVII (the “Indemnitee”) shall promptly notify the other Party (the “Indemnitor”) in writing of any Third Party Claim, in respect of which the Indemnitee intends to claim such indemnification, and the Indemnitor shall have sole control of the defense and/or settlement thereof. The indemnity arrangement in this Section 17.1, 17.2 and 17.3 shall not apply to amounts paid in settlement of any action with respect to a Third Party Claim, if such settlement is effected without the consent of the Indemnitor, which consent shall not be withheld or delayed unreasonably. The failure to deliver written notice to the Indemnitor within a reasonable time after the commencement of any action with respect to a Third Party Claim shall only relieve the Indemnitor of its indemnification obligations under this Article XVII if and to the extent the Indemnitor is actually prejudiced thereby. The Indemnitee shall cooperate fully with the Indemnitor and its legal representatives in the investigation of any action with respect to a Third Party Claim covered by this indemnification.
17.4. Recalls.
(a) Voluntary and Mandatory Recalls: Decision-Making. To the extent that: (i) any Regulatory Authority in the GSK Territory issues a directive or order or requests that the
CONFIDENTIAL
Product be recalled or withdrawn; (ii) a court of competent jurisdiction orders a recall or withdrawal of the Product in the GSK Territory or (iii) GSK, after consultation with Sepracor, determines that an event, incident or circumstance has occurred that means that the Product should be recalled or withdrawn voluntarily in the GSK Territory, the Parties shall recall or withdraw the Product as set forth in this Section 17.4. As between the Parties, GSK shall control and coordinate all activities that GSK deems reasonably necessary in connection with such recall or withdrawal of the Product in the GSK Territory, including making all contact with relevant Regulatory Authorities; provided, however, that GSK shall not take any action with respect to any such recall without first notifying Sepracor in writing, and to the extent practical, consulting in good faith with Sepracor. GSK shall consider in good faith any comments of Sepracor in connection with any aspect of the management of any such recall. For clarity, all matters relating to a withdrawal or recall of the Product in the Sepracor Territory shall be determined, controlled and coordinated solely by Sepracor.
(b) Costs of Recall. All actual direct and documented out-of-pocket expenses for the execution of any recall or withdrawal of the Product (including the cost of any investigation leading to the decision to recall) (“Recall Costs”) pursuant to Section 17.4(a), above, shall initially be shared equally between the Parties; provided that, in each case, responsibility for the Recall Costs shall be subject to the final allocation between the Parties as set out in paragraphs (i) to (iii) below. For clarity, Recall Costs do not include any lost or refunded sales. In the event that it is finally determined, or agreed between the Parties, that such recall or withdrawal is caused by:
(i) breach of Sepracor’s representations, warranties or covenants as set forth in this Agreement, including failure to supply Bulk Tablets conforming to the standards set forth in the Supply Agreement or Quality Agreement, or the gross negligence or willful misconduct of Sepracor or Sepracor’s failure to comply with applicable laws and regulations including cGMP (to be defined in the Quality Agreement) (collectively, the “Fault of Sepracor”), Sepracor shall be responsible for all Recall Costs;
(ii) failure of GSK, or its Affiliates or Sublicensees, to handle, store, package, transport, distribute or use the Bulk Tablets supplied by Sepracor or Product in accordance with applicable laws and regulations or the terms of the applicable Marketing Approval, the Supply Agreement, the Quality Agreement or the gross negligence or willful misconduct of GSK, its Affiliates or Sublicensees, (collectively, “Fault of GSK”), GSK shall be responsible for all Recall Costs; and
(iii) in all other cases, the Recall Costs shall be borne equally by Sepracor and GSK.
17.5. Product Liability Claims.
(a) Notification to the Other. Each Party shall notify the other Party as promptly as practicable if any Third Party Claim is commenced or threatened against such Party alleging Product liability, Product defect, design, packaging or labeling defect, failure to warn
CONFIDENTIAL
or any similar action relating to the use or safety of the Product sold by or under authority of GSK in the GSK Territory (the “Product Liability Claim”).
(b) Cooperation, Counsel and Control. Each Party shall cooperate with the other Party in connection with any such Product Liability Claim that is commenced or threatened against the other Party. If a Product Liability Claim is asserted against both Parties, each Party will have the right to designate counsel to defend itself in the Product Liability Claim. If a Product Liability Claim is brought against one Party but not the other Party, the named Party shall control the defense and/or settlement thereof at its own expense with counsel of its choice, subject to this Section 17.5. In such case, the other Party may participate in the defense and/or settlement thereof at its own expense with counsel of its choice. In any event, the Party that is subject to a Product Liability Claim (if not asserted against both Parties) agrees to keep the other Party hereto informed of all material developments in connection with any such Product Liability Claim.
(c) Settlement, Admissions and Asserting Positions. Neither Party shall settle any Product Liability Claim, or make any admissions or assert any position in such Product Liability Claim, in a manner that would adversely affect the other Party, the Product or the development, manufacture, use or sale thereof without the prior written consent of the other Party, which shall not be unreasonably withheld or delayed.
(d) Bearing the Liabilities. To the extent a Product Liability Claim is caused by: (i) the Fault of Sepracor, Sepracor shall bear all Liabilities from such Product Liability Claim to the extent of its fault; (ii) the Fault of GSK, GSK shall bear all Liabilities from such a Product Liability Claim to the extent of its fault or (iii) circumstances other than those described in Sub-section (i) or (ii), above, the Parties shall share equally the Liabilities from such Product Liability Claim.
ARTICLE XVIII.
DISPUTE RESOLUTION
18.1. Dispute Resolution Process. The Parties recognize that disputes as to certain matters may from time to time arise during the Term of this Agreement that relate to a Party’s rights and/or obligations hereunder. If the Parties cannot resolve any such dispute within thirty (30) days after written notice of a dispute from one Party to another, any Party may, by written notice to the other Party, have such dispute referred to the Senior Executives. The Senior Executives shall negotiate in good faith to resolve the dispute within thirty (30) days. During such period of negotiations, any applicable time periods under this Agreement shall be tolled. If the Senior Executives are unable to resolve the dispute within such time period, either Party may pursue any remedy available to such Party at law or in equity, subject to the terms and conditions of this Agreement and the other agreements expressly contemplated hereunder.
18.2. Governing Law; Litigation; Exclusive Venue. This Agreement and all questions regarding its existence, validity, interpretation, breach or performance of this
Agreement, shall be governed by, and construed and enforced in accordance with, the laws of the State of New York, United States, without reference to its conflicts of law principles. If any dispute cannot be resolved by, and subject to the exhaustion of the procedure set out in Section 18.1, above, any dispute shall be finally settled by litigation brought solely in a United States Federal Court of competent jurisdiction (or state court if no Federal Court has jurisdiction) located in the State of New York, United States, and the Parties hereby submit to the exclusive jurisdiction of such courts.
ARTICLE XIX.
GENERAL PROVISIONS
19.1. Intervening Events. If the performance of any part of this Agreement by either Party is prevented, restricted, interfered with or delayed by any reason or cause beyond the reasonable control of such Party (including: fire, flood, embargo, power shortage or failure, acts of war, insurrection, riot, terrorism, strike, lockout or other labor disturbance, acts of God or any acts, omissions or delays in acting of the other Party) (an “Intervening Event”), the Party so affected shall, upon giving written notice to the other Party, be excused from such performance to the extent of such Intervening Event, provided that the affected Party shall use its substantial efforts to avoid or remove such causes of non-performance and shall continue performance with the utmost dispatch whenever such causes are removed.
(a) Notification of the Other. If either Party becomes aware that such an Intervening Event has occurred, is imminent or likely, it will immediately notify the other;
(b) Efforts to Overcome. The Party which is subject to such Intervening Event shall exert all reasonable efforts to overcome it;
(c) Keeping the Other Informed. Such Party will keep the other informed as to the progress of overcoming it; and
19.2. Waiver of Breach. The failure of either Party at any time or times to require performance of any provision of this Agreement shall in no manner affect its rights at a later time to enforce such rights. No waiver by either Party of any condition or term in any one or more instances shall be construed as a further or continuing waiver of such condition or term or of another condition or term.
19.3. Performance by Affiliates. To the extent that this Agreement imposes obligations on Affiliates of a Party, such Party agrees to cause its Affiliates to perform such obligation. Either Party may use one or more of its Affiliates to perform its obligation hereunder, provided that the Parties will remain liable hereunder for the prompt payment and performance of all their respective obligations hereunder.
19.4. Performance by Subcontractors. Either Party may perform any of its obligations or exercise any of its rights under this Agreement through subcontractors. Such
Party shall ensure that all of its subcontractors comply with, and perform its obligations in accordance with, the terms of this Agreement; provided, however, that the use of such subcontractors by such Party shall not relieve such Party of its respective obligations under this Agreement.
19.5. Modification. No amendment or modification of any provision of this Agreement shall be effective unless in a prior writing signed by both Parties hereto. No provision of this Agreement shall be varied, contradicted or explained by any oral agreement, course of dealing or performance or any other matter not set forth in an agreement in writing and signed by both Parties hereto.
19.6. Severability. In the event any provision of this Agreement should be held invalid, illegal or unenforceable in any jurisdiction, the Parties shall negotiate, in good faith and enter into a valid, legal and enforceable substitute provision that most nearly reflects the original intent of the Parties. All other provisions of this Agreement shall remain in full force and effect in such jurisdiction. Such invalidity, illegality or unenforceability shall not affect the validity, legality or enforceability of such provision in any other jurisdiction.
19.7. Entire Agreement. This Agreement (including the Exhibits and Schedules attached hereto) and the Supply Agreement, Pharmacovigilance Agreement, and Quality Agreement constitutes the entire agreement between the Parties relating to the subject matter hereof and supersedes and cancels all previous express or implied agreements and understandings, negotiations, writings and commitments, either oral or written, in respect to the subject matter hereof. Each of the parties acknowledges and agrees that in entering into this Agreement, and the documents referred to in it, it does not rely on, and shall have no remedy in respect of, any statement, representation, warranty or understanding (whether negligently or innocently made) of any person (whether party to this Agreement or not) other than as expressly set out in this Agreement. Nothing in this clause shall, however, operate to limit or exclude any liability for fraud.
19.8. Language. The language of this Agreement and all activities to be pursued under this Agreement is English. Any and all documents proffered by one Party to the other in fulfillment of any provision of this Agreement shall only be in compliance if in English. Any translation of this Agreement in another language shall be deemed for convenience only and shall never prevail over the original English version. This Agreement is established in the English language.
19.9. Notices. Unless otherwise agreed by the Parties in writing or specified in this Agreement, all communications between the Parties relating to, and all written documentation to be prepared and provided under, this Agreement shall be in the English language. Any notice required or permitted under this Agreement shall be in writing in the English language, delivered personally, sent by facsimile (and promptly confirmed by personal delivery, registered or certified mail or overnight courier), sent by internationally-recognized courier or sent by registered or certified mail, postage prepaid to the following addresses of the
CONFIDENTIAL
Parties (or such other address for a Party as may be at any time thereafter specified by like notice):
|
To Sepracor: |
|
To GSK: |
|
|
|
|
|
|
|
Sepracor Inc. |
|
Glaxo Group Limited |
|
|
00 Xxxxxxxxx Xxxxx |
|
Xxxxx Xxxxxxxx House |
|
|
Marlborough, MA 01752-7010 |
|
Berkeley Avenue |
|
|
USA |
|
Greenford |
|
|
Telephone: |
+ 1- 508-481- 6700 |
|
Middlesex |
|
Facsimile: |
+ 0-000-000-0000 |
|
XX0 0XX |
|
Attention: |
Xxxxxx Xxxxx |
|
UK |
|
|
President & CEO |
|
Facsimile: + 44-20-8047- 6905 |
|
|
|
Attention: Company Secretary |
|
|
|
|
|
|
|
with a copy to: |
|
with a copy to: |
|
|
|
|
|
|
|
Sepracor Inc. |
|
GlaxoSmithKline |
|
|
00 Xxxxxxxxx Xxxxx |
|
000 Xxxxx Xxxx Xxxx |
|
|
Xxxxxxxxxxx, XX 00000-0000 |
|
Xxxxxxxxx |
|
|
XXX |
|
Middlesex |
|
|
Telephone: + 1- 508-481- 6700 |
|
TW8 9GS, UK. |
|
|
Facsimile: + 1-508-357- 7506 |
|
Facsimile: + 44-20-8047- 6897 |
|
|
Attention: Xxxxxx X. Xxxxx |
|
Attention: Legal Operations, |
|
|
Executive Vice President, General |
|
Business Development |
|
|
Counsel and Corporate Secretary |
|
Transactions |
|
Any such notice shall be deemed to have been given: (a) when delivered if personally delivered; (b) on the next Business Day after dispatch if sent by facsimile or by internationally-recognized overnight courier; and/or (c) on the fifth (5th) Business Day following the date of mailing if sent by mail or other internationally-recognized courier. Notices hereunder will not be deemed sufficient if provided only between or among each Party’s representatives on the JSC.
19.10. Assignment. This Agreement shall not be assignable or otherwise transferred, nor may any right or obligations hereunder be assigned or transferred, by either Party to any Third Party without the prior written consent of the other Party; except either Party may assign this Agreement without the consent of the other Party to an entity that acquires all or a substantial part of the business or assets of the assigning Party, whether by merger, acquisition or otherwise, provided that the acquiring Party assumes this Agreement in writing or by operation of law. In addition, either Party shall have the right to assign this Agreement to an Affiliate upon written notice to the non-assigning Party; provided, however, the assigning Party hereby guarantees the performance of this Agreement by such Affiliate. Subject to the foregoing, this
CONFIDENTIAL
Agreement shall inure to the benefit of each Party, its successors and permitted assigns. Any assignment of this Agreement in contravention of this Section 19.10 shall be null and void.
19.11. No Partnership or Joint Venture. Nothing in this Agreement or any action which may be taken pursuant to its terms is intended, or shall be deemed, to establish a joint venture or partnership between GSK and Sepracor. Neither Party to this Agreement shall have any express or implied right or authority to assume or create any obligations on behalf of, or in the name of, the other Party, or to bind the other Party to any contract, agreement or undertaking with any Third Party.
19.12. Interpretation. The captions to the several Articles and Sections of this Agreement are not a part of this Agreement but are included for convenience of reference and shall not affect its meaning or interpretation. In this Agreement: (a) the word “including” shall be deemed to be followed by the phrase “without limitation” or like expression; (b) the singular shall include the plural and vice versa; and (c) masculine, feminine and neuter pronouns and expressions shall be interchangeable. Each accounting term used herein that is not specifically defined herein shall have the meaning given to it under generally accepted accounting principles in the International Financial Reporting Standards consistently applied, but only to the extent consistent with its usage and the other definitions in this Agreement.
19.13. Counterparts. This Agreement may be executed in any number of counterparts each of which shall be deemed an original, and all of which together shall constitute one and the same instrument.
ARTICLE XX.
COMPLIANCE WITH LAW
20.1. Export Laws. Notwithstanding anything to the contrary contained herein, all obligations of Sepracor and GSK are subject to prior compliance with export and import regulations and such other laws and regulations in effect in such jurisdictions or any other relevant country as may be applicable, and to obtaining all necessary approvals required by the applicable agencies of the governments of any relevant countries. Sepracor and GSK shall cooperate with each other and shall provide assistance to the other as reasonably necessary to obtain any required approvals.
20.2. Securities Laws. Each of the Parties acknowledges that it is aware that the securities laws of the United States and the securities laws of other countries prohibit any person who has material non-public information about a publicly listed company from purchasing or selling securities of such company or from communicating such information to any person under circumstances in which it is reasonably foreseeable that such person is likely to purchase or sell such securities. Each Party agrees to comply with such securities laws make its Affiliates,
CONFIDENTIAL
Sublicensees, employees and agents aware of the existence of such securities laws and their need to comply with such laws.
20.3. Certain Payments. Each of the Parties acknowledges that it is aware that the United States and other countries have stringent laws which prohibit persons directly or indirectly to make unlawful payments to, and for the benefit of, government officials and related parties to secure approvals or permission for their activities. Each Party agrees that it will make no such prohibited payments, it will not indirectly make or have made such payments and it will make its Affiliates, employees and agents aware of the existence of such laws and their need to comply with such laws.
20.4. Conduct of Activities. As to all matters contained in this Agreement, each Party shall conduct the activities allocated to it in compliance in all material respects with all applicable laws, rules and regulations and in accordance with good scientific, clinical and manufacturing practices, applicable under the laws and regulations of the country in which such activities are conducted.
CONFIDENTIAL
IN WITNESS WHEREOF, Sepracor Inc. and Glaxo Group Limited, have executed this Development, License and Commercialization Agreement as of the Effective Date.
Sepracor Inc.
|
|
BY: |
/s/ Xxxxxx Xxxxx |
|
Date: |
9/10/07 |
|
|
|
|
|
|
|||||
|
|
NAME: Mr. Xxxxxx Xxxxx |
|
|||||
|
|
|
|
|||||
|
|
TITLE: President & CEO |
|
|||||
|
|
|
|
|||||
|
|
|
|
|||||
|
Glaxo Group Limited |
|
||||||
|
|
|
|
|||||
|
|
BY: |
/s/ Xxxx Xxxxxxxxxx |
|
Date: |
|
|
|
|
|
|
|
|||||
|
|
NAME: Xxxx Xxxxxxxxxx on behalf of |
|
|||||
|
|
|
|
|||||
|
|
Edinburgh Pharmaceutical Industries Limited |
|
|||||
|
|
|
|
|||||
|
|
TITLE: Corporate Director |
|
|||||
CONFIDENTIAL
Schedules and Exhibits
|
Schedules |
|
|
|
|
|
|
|
Schedule 1.39 |
|
Trademarks |
|
Part A: |
Product Trademarks |
|
|
Part B: |
Other Trademarks |
|
|
Schedule 1.44 |
|
Sepracor Patents |
|
Schedule 1.50 |
|
FDA-approved label |
|
Schedule 7.3(c) |
|
Hypothetical Royalty Calculation |
|
Schedule 7.3(e) |
|
Hypothetical Calculation of Cost of Bulk Tablets and Net Sales |
|
Schedule 10.2 |
|
Heads of Terms for Supply Agreement |
|
Schedule 16.2(g) |
|
Threatened and Pending Litigation in GSK Territory |
CONFIDENTIAL
SCHEDULE 1.39
TRADEMARKS
Part A: Product Trademarks
|
Jurisdiction |
|
Trademark |
|
Status |
|
App. No. |
|
|
|
Reg. Date |
||
|
Brazil |
|
LUNIVIA |
|
Published |
|
825757444 |
|
11-Aug-2003 |
|
|
|
|
|
Chile |
|
LUNIVIA |
|
Registered |
|
617060 |
|
08-Aug-2003 |
|
787236 |
|
00-Xxx-0000 |
|
Xxxxx |
|
LUNIVIA |
|
Registered |
|
3516600 |
|
07-Apr-2003 |
|
3516600 |
|
00-Xxx-0000 |
|
Xxxxxxxx |
|
LUNIVIA |
|
Registered |
|
|
|
|
|
296477 |
|
27-Apr-2004 |
|
European Community |
|
LUNIVIA |
|
Registered |
|
3077088 |
|
10-Mar-2003 |
|
3077088 |
|
23-Nov-2004 |
|
Hong Kong |
|
LUNIVIA |
|
Registered |
|
300003040 |
|
07-Apr-2003 |
|
300003040 |
|
06-Oct-2003 |
|
Indonesia |
|
LUNIVIA |
|
Registered |
|
|
|
07-Apr-2003 |
|
IDM000029089 |
|
04-Feb-2005 |
|
Xxxx |
|
XXXXXXX |
|
Xxxxxxxxxx |
|
|
|
|
|
00000 |
|
00-Xxx-0000 |
|
Xxxxxxx |
|
LUNIVIA |
|
Pending |
|
20070930 |
|
00-Xxx-0000 |
|
|
|
|
|
Xxxxxx Xxxxxxx |
|
LUNIVIA |
|
Published |
|
2449182 |
|
12-Mar-2007 |
|
|
|
|
|
Ireland |
|
LUNIVIA |
|
Published |
|
235997 |
|
00-Xxx-0000 |
|
|
|
|
|
Xxxxxx |
|
LUNIVIA |
|
Pending |
|
708067 |
|
14-Mar-2007 |
|
|
|
|
|
Korea |
|
LUNIVIA |
|
Pending |
|
25997 |
|
14-May-2007 |
|
|
|
|
|
Venezuela |
|
LUNIVIA |
|
Registered |
|
11085 |
|
11-Nov-2005 |
|
P264537 |
|
00-Xxx-0000 |
|
Xxxxxxxxxxx |
|
LUNIVIA |
|
Registered |
|
01636 |
|
18-Mar-2003 |
|
510653 |
|
22-May-2003 |
|
Taiwan |
|
LUNIVIA |
|
Registered |
|
92017207 |
|
10-Apr-2003 |
|
1074103 |
|
00-Xxx-0000 |
|
Xxxxxxxx |
|
LUNIVIA |
|
Registered |
|
515824 |
|
01-Aug-2002 |
|
KOR2215802 |
|
10-Apr-2003 |
CONFIDENTIAL
SCHEDULE 1.39
TRADEMARKS
Part B: Other Trademarks
None
CONFIDENTIAL
SCHEDULE 1.44
|
COUNTRY |
|
PATENT NO. |
|
FILING DATE |
|
Argentina |
|
248024 |
|
16 Jan. 0000 |
|
Xxxxxxxxx |
|
671797 |
|
16 Jan. 0000 |
|
Xxxxxxx |
|
121089 |
|
16 Jan. 1992 |
|
Bangladesh |
|
1002434 |
|
15 Jan. 0000 |
|
Xxxxxxx |
|
1854 |
|
16 Jan. 0000 |
|
Xxxxxxx |
|
EP609210 X0 |
|
00 Xxx. 0000 |
|
Xxxxx Xxxxxxxx |
|
281011 |
|
16 Jan. 1992 |
|
Denmark |
|
EP609210 X0 |
|
00 Xxx. 0000 |
|
Xxxxxxx |
|
100331 |
|
16 Jan. 1992 |
|
France |
|
EP609210 B1 |
|
16 Jan. 1992 |
|
Germany |
|
EP609210 B1 |
|
16 Jan. 1992 |
|
Great Britain |
|
EP609210 X0 |
|
00 Xxx. 0000 |
|
Xxxxxx |
|
EP609210 B1 |
|
16 Jan. 0000 |
|
Xxxxxxx |
|
218928 |
|
16 Jan. 0000 |
|
Xxxxxxx |
|
66110 |
|
16 Jan. 1992 |
|
Israel |
|
100677 |
|
16 Jan. 0000 |
|
Xxxxx |
|
EP609210 B1 |
|
16 Jan. 1992 |
|
Luxembourg |
|
EP609210 B1 |
|
16 Jan. 1992 |
|
Morocco |
|
22392 |
|
15 Jan. 0000 |
|
Xxxxxxxxxxx |
|
EP609210 X0 |
|
00 Xxx. 0000 |
|
Xxx Xxxxxxx |
|
241313 |
|
16 Jan. 1992 |
|
Nigeria |
|
11252 |
|
16 Jan. 1992 |
|
Norway |
|
EP609210 B1 |
|
16 Jan. 1992 |
|
Pakistan |
|
132928 |
|
16 Jan. 0000 |
|
Xxxxxxxxxxx |
|
30982 |
|
16 Jan. 0000 |
|
Xxxxxx |
|
166976 |
|
16 Jan. 1992 |
|
Portugal |
|
EP609210 B1 |
|
16 Jan. 1992 |
|
Russia |
|
2110519 |
|
16 Jan. 0000 |
|
Xxxxxx |
|
48878 |
|
17 Jan. 0000 |
|
Xxxxxxxx |
|
279060 |
|
16 Jan. 0000 |
|
Xxxxx Xxxxxx |
|
92302 |
|
15 Jan. 0000 |
|
Xxxxx |
|
2071486 |
|
16 Jan. 1992 |
|
Sweden |
|
EP609210 B1 |
|
16 Jan. 0000 |
|
Xxxxxxxxxxx |
|
EP609210 B1 |
|
16 Jan. 1992 |
|
Tunisia |
|
16531 |
|
17 Jan. 1992 |
CONFIDENTIAL
SCHEDULE 1.50
FDA-APPROVED LABEL
(Attached)
|
Rx only |
|
C-IV |
LUNESTA® (eszopiclone) TABLETS
1 mg, 2 mg, 3 mg
PRESCRIBING INFORMATION
LUNESTA (eszopiclone) is a nonbenzodiazepine hypnotic agent that is a pyrrolopyrazine derivative of the cyclopyrrolone class. The chemical name of eszopiclone is (+)-(5S)-6-(5-chloropyridin-2-yl)-7-oxo-6,7-dihydro-5H-pyrrolo[3,4-b] pyrazin-5-yl 4-methylpiperazine-1-carboxylate. Its molecular weight is 388.81, and its empirical formula is C17H17ClN6O3. Eszopiclone has a single chiral center with an (S)-configuration. It has the following chemical structure:
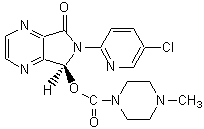
Eszopiclone is a white to light-yellow crystalline solid. Eszopiclone is very slightly soluble in water, slightly soluble in ethanol, and soluble in phosphate buffer (pH 3.2).
Eszopiclone is formulated as film-coated tablets for oral administration. LUNESTA tablets contain 1 mg, 2 mg, or 3 mg eszopiclone and the following inactive ingredients: calcium phosphate, colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, titanium dioxide, and triacetin. In addition, both the 1 mg and 3 mg tablets contain FD&C Blue #2.
CLINICAL PHARMACOLOGY:
Pharmacodynamics
The precise mechanism of action of eszopiclone as a hypnotic is unknown, but its effect is believed to result from its interaction with GABA-receptor complexes at binding domains located close to or allosterically coupled to benzodiazepine receptors. Eszopiclone is a nonbenzodiazepine hypnotic that is a pyrrolopyrazine derivative of the cyclopyrrolone class with a chemical structure unrelated to pyrazolopyrimidines, imidazopyridines, benzodiazepines, barbiturates, or other drugs with known hypnotic properties.
1
Pharmacokinetics
The pharmacokinetics of eszopiclone have been investigated in healthy subjects (adult and elderly) and in patients with hepatic disease or renal disease. In healthy subjects, the pharmacokinetic profile was examined after single doses of up to 7.5 mg and after once-daily administration of 1, 3, and 6 mg for 7 days. Eszopiclone is rapidly absorbed, with a time to peak concentration (tmax) of approximately 1 hour and a terminal-phase elimination half-life (t1/2) of approximately 6 hours. In healthy adults, LUNESTA does not accumulate with once-daily administration, and its exposure is dose-proportional over the range of 1 to 6 mg.
Absorption And Distribution
Eszopiclone is rapidly absorbed following oral administration. Peak plasma concentrations are achieved within approximately 1 hour after oral administration. Eszopiclone is weakly bound to plasma protein (52-59%). The large free fraction suggests that eszopiclone disposition should not be affected by drug-drug interactions caused by protein binding. The blood-to-plasma ratio for eszopiclone is less than one, indicating no selective uptake by red blood cells.
Metabolism
Following oral administration, eszopiclone is extensively metabolized by oxidation and demethylation. The primary plasma metabolites are (S)-zopiclone-N-oxide and (S)-N-desmethyl zopiclone; the latter compound binds to GABA receptors with substantially lower potency than eszopiclone, and the former compound shows no significant binding to this receptor. In vitro studies have shown that CYP3A4 and CYP2E1 enzymes are involved in the metabolism of eszopiclone. Eszopiclone did not show any inhibitory potential on CYP450 1A2, 2A6, 2C9, 2C19, 2D6, 2E1, and 3A4 in cryopreserved human hepatocytes.
Elimination
After oral administration, eszopiclone is eliminated with a mean t1/2 of approximately 6 hours. Up to 75% of an oral dose of racemic zopiclone is excreted in the urine, primarily as metabolites. A similar excretion profile would be expected for eszopiclone, the S-isomer of racemic zopiclone. Less than 10% of the orally administered eszopiclone dose is excreted in the urine as parent drug.
Effect Of Food
In healthy adults, administration of a 3 mg dose of eszopiclone after a high-fat meal resulted in no change in AUC, a reduction in mean Cmax of 21%, and delayed tmax by approximately 1 hour. The half-life remained unchanged, approximately 6 hours. The effects of LUNESTA on sleep onset may be reduced if it is taken with or immediately after a high-fat/heavy meal.
2
Special Populations
Age
Compared with non-elderly adults, subjects 65 years and older had an increase of 41% in total exposure (AUC) and a slightly prolonged elimination of eszopiclone (t1/2 approximately 9 hours). Cmax was unchanged. Therefore, in elderly patients the starting dose of LUNESTA should be decreased to 1 mg and the dose should not exceed 2 mg.
Gender
The pharmacokinetics of eszopiclone in men and women are similar.
Race
In an analysis of data on all subjects participating in Phase 1 studies of eszopiclone, the pharmacokinetics for all races studied appeared similar.
Hepatic Impairment
Pharmacokinetics of a 2 mg eszopiclone dose were assessed in 16 healthy volunteers and in 8 subjects with mild, moderate, and severe liver disease. Exposure was increased 2-fold in severely impaired patients compared with the healthy volunteers. Cmax and tmax were unchanged. The dose of LUNESTA should not be increased above 2 mg in patients with severe hepatic impairment. No dose adjustment is necessary for patients with mild-to-moderate hepatic impairment. LUNESTA should be used with caution in patients with hepatic impairment. (See Dosage AND Administration.)
Renal Impairment
The pharmacokinetics of eszopiclone were studied in 24 patients with mild, moderate, or severe renal impairment. AUC and Cmax were similar in the patients compared with demographically matched healthy control subjects. No dose adjustment is necessary in patients with renal impairment, since less than 10% of the orally administered eszopiclone dose is excreted in the urine as parent drug.
Drug Interactions
Eszopiclone is metabolized by CYP3A4 and CYP2E1 via demethylation and oxidation. There were no pharmacokinetic or pharmacodynamic interactions between eszopiclone and paroxetine, digoxin, or warfarin. When eszopiclone was coadministered with olanzapine, no pharmacokinetic interaction was detected in levels of eszopiclone or olanzapine, but a pharmacodynamic interaction was seen on a measure of psychomotor function. Eszopiclone and lorazepam decreased each other’s Cmax by 22%. Coadministration of eszopiclone 3 mg to subjects receiving ketoconazole 400 mg, a potent inhibitor of CYP3A4, resulted in a 2.2-fold
3
increase in exposure to eszopiclone. LUNESTA would not be expected to alter the clearance of drugs metabolized by common CYP450 enzymes. (See PRECAUTIONS.)
Clinical Trials:
The effect of LUNESTA on reducing sleep latency and improving sleep maintenance was established in studies with 2100 subjects (ages 18-86) with chronic and transient insomnia in six placebo-controlled trials of up to 6 months’ duration. Two of these trials were in elderly patients (n=523). Overall, at the recommended adult dose (2-3 mg) and xxxxxxx xxxx (0-0 xx), XXXXXXX significantly decreased sleep latency and improved measures of sleep maintenance (objectively measured as wake time after sleep onset [WASO] and subjectively measured as total sleep time).
Transient Insomnia
Healthy adults were evaluated in a model of transient insomnia (n=436) in a sleep laboratory in a double-blind, parallel-group, single-night trial comparing two doses of eszopiclone and placebo. LUNESTA 3 mg was superior to placebo on measures of sleep latency and sleep maintenance, including polysomnographic (PSG) parameters of latency to persistent sleep (LPS) and WASO.
Chronic Insomnia (Adults And Elderly)
The effectiveness of LUNESTA was established in five controlled studies in chronic insomnia. Three controlled studies were in adult subjects, and two controlled studies were in elderly subjects with chronic insomnia.
Adults
In the first study, adults with chronic insomnia (n=308) were evaluated in a double-blind, parallel-group trial of 6 weeks’ duration comparing LUNESTA 2 mg and 3 mg with placebo. Objective endpoints were measured for 4 weeks. Both 2 mg and 3 mg were superior to placebo on LPS at 4 weeks. The 3 mg dose was superior to placebo on WASO.
In the second study, adults with chronic insomnia (n=788) were evaluated using subjective measures in a double-blind, parallel-group trial comparing the safety and efficacy of LUNESTA 3 mg with placebo administered nightly for 6 months. LUNESTA was superior to placebo on subjective measures of sleep latency, total sleep time, and WASO.
In addition, a 6-period cross-over PSG study evaluating eszopiclone doses of 1 to 3 mg, each given over a 2-day period, demonstrated effectiveness of all doses on LPS, and of 3 mg on WASO. In this trial, the response was dose-related.
4
Elderly
Elderly subjects (ages 65-86) with chronic insomnia were evaluated in two double-blind, parallel-group trials of 2 weeks’ duration. One study (n=231) compared the effects of LUNESTA with placebo on subjective outcome measures, and the other (n=292) on objective and subjective outcome measures. The first study compared 1 mg and 2 mg of LUNESTA with placebo, while the second study compared 2 mg of LUNESTA with placebo. All doses were superior to placebo on measures of sleep latency. In both studies, 2 mg of LUNESTA was superior to placebo on measures of sleep maintenance.
Studies Pertinent To Safety Concerns For Sedative/Hypnotic Drugs
Cognitive, Memory, Sedative, and Psychomotor Effects
In two double-blind, placebo-controlled, single-dose cross-over studies of 12 patients each (one study in patients with insomnia; one in normal volunteers), the effects of LUNESTA 2 and 3 mg were assessed on 20 measures of cognitive function and memory at 9.5 and 12 hours after a nighttime dose. Although results suggested that patients receiving LUNESTA 3 mg performed more poorly than patients receiving placebo on a very small number of these measures at 9.5 hours post-dose, no consistent pattern of abnormalities was seen.
In a 6-month double-blind, placebo-controlled trial of nightly administered LUNESTA 3 mg, 8/593 subjects treated with LUNESTA 3 mg (1.3%) and 0/195 subjects treated with placebo (0%) spontaneously reported memory impairment. The majority of these events were mild in nature (5/8), and none were reported as severe. Four of these events occurred within the first 7 days of treatment and did not recur. The incidence of spontaneously reported confusion in this 6-month study was 0.5% in both treatment arms. In a 6-week adult study of nightly administered LUNESTA 2 mg or 3 mg or placebo, the spontaneous reporting rates for confusion were 0%, 3.0%, and 0%, respectively, and for memory impairment were 1%, 1%, and 0%, respectively.
In a 2-week study of 264 elderly insomniacs randomized to either nightly LUNESTA 2 mg or placebo, spontaneous reporting rates of confusion and memory impairment were 0% vs. 0.8% and 1.5% vs. 0%, respectively. In another 2-week study of 231 elderly insomniacs, the spontaneous reporting rates for the 1 mg, 2 mg, and placebo groups for confusion were 0%, 2.5%, and 0%, respectively, and for memory impairment were 1.4%, 0%, and 0%, respectively.
A study of normal subjects exposed to single fixed doses of LUNESTA from 1 to 7.5 mg using the DSST to assess sedation and psychomotor function at fixed times after dosing (hourly up to 16 hours) found the expected sedation and reduction in psychomotor function. This was maximal at 1 hour and present up to 4 hours, but was no longer present by 5 hours.
In another study, patients with insomnia were given 2 or 3 mg doses of LUNESTA nightly, with DSST assessed on the mornings following days 1, 15, and 29 of treatment. While both the placebo and LUNESTA 3 mg groups showed an improvement in DSST scores relative to baseline the following morning (presumably due to a learning effect), the improvement in the
5
placebo group was greater and reached statistical significance on night 1, although not on nights 15 and 29. For the LUNESTA 2 mg group, DSST change scores were not significantly different from placebo at any time point.
Withdrawal-Emergent Anxiety And Insomnia
During nightly use for an extended period, pharmacodynamic tolerance or adaptation has been observed with other hypnotics. If a drug has a short elimination half-life, it is possible that a relative deficiency of the drug or its active metabolites (i.e., in relationship to the receptor site) may occur at some point in the interval between each night’s use. This is believed to be responsible for two clinical findings reported to occur after several weeks of nightly use of other rapidly eliminated hypnotics: increased wakefulness during the last quarter of the night and the appearance of increased signs of daytime anxiety.
In a 6-month double-blind, placebo-controlled study of nightly administration of LUNESTA 3 mg, rates of anxiety reported as an adverse event were 2.1% in the placebo arm and 3.7% in the LUNESTA arm. In a 6-week adult study of nightly administration, anxiety was reported as an adverse event in 0%, 2.9%, and 1.0% of the placebo, 2 mg, and 3 mg treatment arms, respectively. In this study, single-blind placebo was administered on nights 45 and 46, the first and second days of withdrawal from study drug. New adverse events were recorded during the withdrawal period, beginning with day 45, up to 14 days after discontinuation. During this withdrawal period, 105 subjects previously taking nightly LUNESTA 3 mg for 44 nights spontaneously reported anxiety (1%), abnormal dreams (1.9%), hyperesthesia (1%), and neurosis (1%), while none of 99 subjects previously taking placebo reported any of these adverse events during the withdrawal period.
Rebound insomnia, defined as a dose-dependent temporary worsening in sleep parameters (latency, sleep efficiency, and number of awakenings) compared with baseline following discontinuation of treatment, is observed with short- and intermediate-acting hypnotics. Rebound insomnia following discontinuation of LUNESTA relative to placebo and baseline was examined objectively in a 6-week adult study on the first 2 nights of discontinuation (nights 45 and 46) following 44 nights of active treatment with 2 mg or 3 mg. In the LUNESTA 2 mg group, compared with baseline, there was a significant increase in WASO and a decrease in sleep efficiency, both occurring only on the first night after discontinuation of treatment. No changes from baseline were noted in the LUNESTA 3 mg group on the first night after discontinuation, and there was a significant improvement in LPS and sleep efficiency compared with baseline following the second night of discontinuation. Comparisons of changes from baseline between LUNESTA and placebo were also performed. On the first night after discontinuation of LUNESTA 2 mg, LPS and WASO were significantly increased and sleep efficiency was reduced; there were no significant differences on the second night. On the first night following discontinuation of LUNESTA 3 mg, sleep efficiency was significantly reduced. No other differences from placebo were noted in any other sleep parameter on either the first or second night following discontinuation. For both doses, the discontinuation-emergent effect was mild, had the characteristics of the return of the symptoms of chronic insomnia, and appeared to resolve by the second night after LUNESTA discontinuation.
6
INDICATIONS AND USAGE:
LUNESTA is indicated for the treatment of insomnia. In controlled outpatient and sleep laboratory studies, LUNESTA administered at bedtime decreased sleep latency and improved sleep maintenance.
The clinical trials performed in support of efficacy were up to 6 months in duration. The final formal assessments of sleep latency and maintenance were performed at 4 weeks in the 6-week study, at the end of both 2-week studies and at the end of the 6-month study.
CONTRAINDICATIONS:
None known.
WARNINGS:
Because sleep disturbances may be the presenting manifestation of a physical and/or psychiatric disorder, symptomatic treatment of insomnia should be initiated only after a careful evaluation of the patient. The failure of insomnia to remit after 7 to 10 days of treatment may indicate the presence of a primary psychiatric and/or medical illness that should be evaluated. Worsening of insomnia or the emergence of new thinking or behavior abnormalities may be the consequence of an unrecognized psychiatric or physical disorder. Such findings have emerged during the course of treatment with sedative/hypnotic drugs, including LUNESTA. Because some of the important adverse effects of LUNESTA appear to be dose-related, it is important to use the lowest possible effective dose, especially in the elderly (see DOSAGE AND ADMINISTRATION).
A variety of abnormal thinking and behavior changes have been reported to occur in association with the use of sedative/hypnotics. Some of these changes may be characterized by decreased inhibition (e.g., aggressiveness and extroversion that seem out of character), similar to effects produced by alcohol and other CNS depressants. Other reported behavioral changes have included bizarre behavior, agitation, hallucinations, and depersonalization. Complex behaviors such as “sleep-driving” (i.e., driving while not fully awake after ingestion of a sedative-hypnotic, with amnesia for the event) have been reported. These events can occur in sedative-hypnotic-naïve as well as in sedative-hypnotic-experienced persons. Although behaviors such as sleep-driving may occur with LUNESTA alone at therapeutic doses, the use of alcohol and other CNS depressants with LUNESTA appears to increase the risk of such behaviors, as does the use of LUNESTA at doses exceeding the maximum recommended dose. Due to the risk to the patient and the community, discontinuation of LUNESTA should be strongly considered for patients who report a “sleep-driving” episode. Other complex behaviors (e.g., preparing and eating food, making phone calls, or having sex) have been reported in patients who are not fully awake after taking a sedative-hypnotic. As with sleep-driving, patients usually do not remember these events. Amnesia and other neuropsychiatric symptoms may occur unpredictably. In primarily
7
depressed patients, worsening of depression, including suicidal thinking, has been reported in association with the use of sedative/hypnotics.
It can rarely be determined with certainty whether a particular instance of the abnormal behaviors listed above are drug-induced, spontaneous in origin, or a result of an underlying psychiatric or physical disorder. Nonetheless, the emergence of any new behavioral sign or symptom of concern requires careful and immediate evaluation.
Following rapid dose decrease or abrupt discontinuation of the use of sedative/hypnotics, there have been reports of signs and symptoms similar to those associated with withdrawal from other CNS-depressant drugs (see Drug Abuse and Dependence).
LUNESTA, like other hypnotics, has CNS-depressant effects. Because of the rapid onset of action, LUNESTA should only be ingested immediately prior to going to bed or after the patient has gone to bed and has experienced difficulty falling asleep. Patients receiving LUNESTA should be cautioned against engaging in hazardous occupations requiring complete mental alertness or motor coordination (e.g., operating machinery or driving a motor vehicle) after ingesting the drug, and be cautioned about potential impairment of the performance of such activities on the day following ingestion of LUNESTA. LUNESTA, like other hypnotics, may produce additive CNS-depressant effects when coadministered with other psychotropic medications, anticonvulsants, antihistamines, ethanol, and other drugs that themselves produce CNS depression. LUNESTA should not be taken with alcohol. Dose adjustment may be necessary when LUNESTA is administered with other CNS-depressant agents, because of the potentially additive effects.
Severe anaphylactic and anaphylactoid reactions
Rare cases of angioedema involving the tongue, glottis or larynx have been reported in patients after taking the first or subsequent doses of sedative-hypnotics, including LUNESTA. Some patients have had additional symptoms such as dyspnea, throat closing, or nausea and vomiting that suggest anaphylaxis. Some patients have required medical therapy in the emergency department. If angioedema involves the tongue, glottis or larynx, airway obstruction may occur and be fatal. Patients who develop angioedema after treatment with LUNESTA should not be rechallenged with the drug.
PRECAUTIONS:
General
Timing Of Drug Administration
LUNESTA should be taken immediately before bedtime. Taking a sedative/hypnotic while still up and about may result in short-term memory impairment, hallucinations, impaired coordination, dizziness, and lightheadedness.
8
Use In The Elderly And/Or Debilitated Patients
Impaired motor and/or cognitive performance after repeated exposure or unusual sensitivity to sedative/hypnotic drugs is a concern in the treatment of elderly and/or debilitated patients. The recommended starting dose of LUNESTA for these patients is 1 mg. (See DOSAGE AND ADMINISTRATION.)
Use In Patients With Concomitant Illness
Clinical experience with eszopiclone in patients with concomitant illness is limited. Eszopiclone should be used with caution in patients with diseases or conditions that could affect metabolism or hemodynamic responses.
A study in healthy volunteers did not reveal respiratory-depressant effects at doses 2.5-fold higher (7 mg) than the recommended dose of eszopiclone. Caution is advised, however, if LUNESTA is prescribed to patients with compromised respiratory function.
The dose of LUNESTA should be reduced to 1 mg in patients with severe hepatic impairment, because systemic exposure is doubled in such subjects. No dose adjustment appears necessary for subjects with mild or moderate hepatic impairment. No dose adjustment appears necessary in subjects with any degree of renal impairment, since less than 10% of eszopiclone is excreted unchanged in the urine.
The dose of LUNESTA should be reduced in patients who are administered potent inhibitors of CYP3A4, such as ketoconazole, while taking LUNESTA. Downward dose adjustment is also recommended when LUNESTA is administered with agents having known CNS-depressant effects.
Use In Patients With Depression
Sedative/hypnotic drugs should be administered with caution to patients exhibiting signs and symptoms of depression. Suicidal tendencies may be present in such patients, and protective measures may be required. Intentional overdose is more common in this group of patients; therefore, the least amount of drug that is feasible should be prescribed for the patient at any one time.
Information For Patients
Patient information is printed at the bottom of this insert. To assure safe and effective use of LUNESTA, this information and the instructions provided in the patient information section should be discussed with patients.
SPECIAL CONCERNS “Sleep-Driving” and other complex behaviors
There have been reports of people getting out of bed after taking a sedative-hypnotic and driving their cars while not fully awake, often with no memory of the event. If a patient experiences
9
such an episode, it should be reported to his or her doctor immediately, since “sleep-driving” can be dangerous. This behavior is more likely to occur when LUNESTA is taken with alcohol or other central nervous system depressants (see WARNINGS). Other complex behaviors (e.g., preparing and eating food, making phone calls, or having sex) have been reported in patients who are not fully awake after taking a sedative-hypnotic. As with sleep-driving, patients usually do not remember these events.
Laboratory Tests
There are no specific laboratory tests recommended.
Drug Interactions
CNS-Active Drugs
Ethanol: An additive effect on psychomotor performance was seen with coadministration of eszopiclone and ethanol 0.70 g/kg for up to 4 hours after ethanol administration.
Paroxetine: Coadministration of single doses of eszopiclone 3 mg and paroxetine 20 mg daily for 7 days produced no pharmacokinetic or pharmacodynamic interaction.
Lorazepam: Coadministration of single doses of eszopiclone 3 mg and lorazepam 2 mg did not have clinically relevant effects on the pharmacodynamics or pharmacokinetics of either drug.
Olanzapine: Coadministration of eszopiclone 3 mg and olanzapine 10 mg produced a decrease in DSST scores. The interaction was pharmacodynamic; there was no alteration in the pharmacokinetics of either drug.
Drugs That Inhibit CYP3A4 (Ketoconazole)
CYP3A4 is a major metabolic pathway for elimination of eszopiclone. The AUC of eszopiclone was increased 2.2-fold by coadministration of ketoconazole, a potent inhibitor of CYP3A4, 400 mg daily for 5 days. Cmax and t1/2 were increased 1.4-fold and 1.3-fold, respectively. Other strong inhibitors of CYP3A4 (e.g., itraconazole, clarithromycin, nefazodone, troleandomycin, ritonavir, nelfinavir) would be expected to behave similarly.
Drugs That Induce CYP3A4 (Rifampicin)
Racemic zopiclone exposure was decreased 80% by concomitant use of rifampicin, a potent inducer of CYP3A4. A similar effect would be expected with eszopiclone.
Drugs Highly Bound To Plasma Protein
Eszopiclone is not highly bound to plasma proteins (52-59% bound); therefore, the disposition of eszopiclone is not expected to be sensitive to alterations in protein binding. Administration of
10
eszopiclone 3 mg to a patient taking another drug that is highly protein-bound would not be expected to cause an alteration in the free concentration of either drug.
Drugs With A Narrow Therapeutic Index
Digoxin: A single dose of eszopiclone 3 mg did not affect the pharmacokinetics of digoxin measured at steady state following dosing of 0.5 mg twice daily for one day and 0.25 mg daily for the next 6 days.
Warfarin: Eszopiclone 3 mg administered daily for 5 days did not affect the pharmacokinetics of (R)- or (S)-warfarin, nor were there any changes in the pharmacodynamic profile (prothrombin time) following a single 25 mg oral dose of warfarin.
Carcinogenesis, Mutagenesis, Impairment Of Fertility
Carcinogenesis
In a carcinogenicity study in Xxxxxxx-Xxxxxx rats in which eszopiclone was given by oral gavage, no increases in tumors were seen; plasma levels (AUC) of eszopiclone at the highest dose used in this study (16 mg/kg/day) are estimated to be 80 (females) and 20 (males) times those in humans receiving the maximum recommended human dose (MRHD). However, in a carcinogenicity study in Xxxxxxx-Xxxxxx rats in which racemic zopiclone was given in the diet, and in which plasma levels of eszopiclone were reached that were greater than those reached in the above study of eszopiclone, an increase in mammary gland adenocarcinomas in females and an increase in thyroid gland follicular cell adenomas and carcinomas in males were seen at the highest dose of 100 mg/kg/day. Plasma levels of eszopiclone at this dose are estimated to be 150 (females) and 70 (males) times those in humans receiving the MRHD. The mechanism for the increase in mammary adenocarcinomas is unknown. The increase in thyroid tumors is thought to be due to increased levels of TSH secondary to increased metabolism of circulating thyroid hormones, a mechanism that is not considered to be relevant to humans.
In a carcinogenicity study in B6C3F1 mice in which racemic zopiclone was given in the diet, an increase in pulmonary carcinomas and carcinomas plus adenomas in females and an increase in skin fibromas and sarcomas in males were seen at the highest dose of 100 mg/kg/day. Plasma levels of eszopiclone at this dose are estimated to be 8 (females) and 20 (males) times those in humans receiving the MRHD. The skin tumors were due to skin lesions induced by aggressive behavior, a mechanism that is not relevant to humans. A carcinogenicity study was also performed in which CD-1 mice were given eszopiclone at doses up to 100 mg/kg/day by oral gavage; although this study did not reach a maximum tolerated dose, and was thus inadequate for overall assessment of carcinogenic potential, no increases in either pulmonary or skin tumors were seen at doses producing plasma levels of eszopiclone estimated to be 90 times those in humans receiving the MRHD — i.e., 12 times the exposure in the racemate study.
Eszopiclone did not increase tumors in a p53 transgenic mouse bioassay at oral doses up to 300 mg/kg/day.
11
Mutagenesis
Eszopiclone was positive in the mouse lymphoma chromosomal aberration assay and produced an equivocal response in the Chinese hamster ovary cell chromosomal aberration assay. It was not mutagenic or clastogenic in the bacterial Xxxx xxxx mutation assay, in an unscheduled DNA synthesis assay, or in an in vivo mouse bone marrow micronucleus assay.
(S)-N-desmethyl zopiclone, a metabolite of eszopiclone, was positive in the Chinese hamster ovary cell and human lymphocyte chromosomal aberration assays. It was negative in the bacterial Xxxx mutation assay, in an in vitro 32P-postlabeling DNA adduct assay, and in an in vivo mouse bone marrow chromosomal aberration and micronucleus assay.
Impairment Of Fertility
Eszopiclone was given by oral gavage to male rats at doses up to 45 mg/kg/day from 4 weeks premating through mating and to female rats at doses up to 180 mg/kg/day from 2 weeks premating through day 7 of pregnancy. An additional study was performed in which only females were treated, up to 180 mg/kg/day. Eszopiclone decreased fertility, probably because of effects in both males and females, with no females becoming pregnant when both males and females were treated with the highest dose; the no-effect dose in both sexes was 5 mg/kg (16 times the MRHD on a mg/m2 basis). Other effects included increased pre-implantation loss (no-effect dose 25 mg/kg), abnormal estrus cycles (no-effect dose 25 mg/kg), and decreases in sperm number and motility and increases in morphologically abnormal sperm (no-effect dose 5 mg/kg).
Pregnancy
Pregnancy Category C
Eszopiclone administered by oral gavage to pregnant rats and rabbits during the period of organogenesis showed no evidence of teratogenicity up to the highest doses tested (250 and 16 mg/kg/day in rats and rabbits, respectively; these doses are 800 and 100 times, respectively, the maximum recommended human dose [MRHD] on a mg/m2 basis). In the rat, slight reductions in fetal weight and evidence of developmental delay were seen at maternally toxic doses of 125 and 150 mg/kg/day, but not at 62.5 mg/kg/day (200 times the MRHD on a mg/m2 basis).
Eszopiclone was also administered by oral gavage to pregnant rats throughout the pregnancy and lactation periods at doses of up to 180 mg/kg/day. Increased post-implantation loss, decreased postnatal pup weights and survival, and increased pup startle response were seen at all doses; the lowest dose tested, 60 mg/kg/day, is 200 times the MRHD on a mg/m2 basis. These doses did not produce significant maternal toxicity. Eszopiclone had no effects on other behavioral measures or reproductive function in the offspring.
There are no adequate and well-controlled studies of eszopiclone in pregnant women. Eszopiclone should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
12
Labor And Delivery
LUNESTA has no established use in labor and delivery.
Nursing Mothers
It is not known whether LUNESTA is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when LUNESTA is administered to a nursing woman.
Pediatric Use
Safety and effectiveness of eszopiclone in children below the age of 18 have not been established.
Geriatric Use
A total of 287 subjects in double-blind, parallel-group, placebo-controlled clinical trials who received eszopiclone were 65 to 86 years of age. The overall pattern of adverse events for elderly subjects (median age = 71 years) in 2-week studies with nighttime dosing of 2 mg eszopiclone was not different from that seen in younger adults (see ADVERSE REACTIONS, Table 2). LUNESTA 2 mg exhibited significant reduction in sleep latency and improvement in sleep maintenance in the elderly population.
Adverse Reactions:
The premarketing development program for LUNESTA included eszopiclone exposures in patients and/or normal subjects from two different groups of studies: approximately 400 normal subjects in clinical pharmacology/pharmacokinetic studies, and approximately 1550 patients in placebo-controlled clinical effectiveness studies, corresponding to approximately 263 patient-exposure years. The conditions and duration of treatment with LUNESTA varied greatly and included (in overlapping categories) open-label and double-blind phases of studies, inpatients and outpatients, and short-term and longer-term exposure. Adverse reactions were assessed by collecting adverse events, results of physical examinations, xxxxx xxxxx, weights, laboratory analyses, and ECGs.
Adverse events during exposure were obtained primarily by general inquiry and recorded by clinical investigators using terminology of their own choosing. Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse events without first grouping similar types of events into a smaller number of standardized event categories. In the tables and tabulations that follow, COSTART terminology has been used to classify reported adverse events.
13
The stated frequencies of adverse events represent the proportion of individuals who experienced, at least once, a treatment-emergent adverse event of the type listed. An event was considered treatment-emergent if it occurred for the first time or worsened while the patient was receiving therapy following baseline evaluation.
Adverse Findings Observed In Placebo-Controlled Trials
Adverse Events Resulting In Discontinuation Of Treatment
In placebo-controlled, parallel-group clinical trials in the elderly, 3.8% of 208 patients who received placebo, 2.3% of 215 patients who received 2 mg LUNESTA, and 1.4% of 72 patients who received 1 mg LUNESTA discontinued treatment due to an adverse event. In the 6-week parallel-group study in adults, no patients in the 3 mg arm discontinued because of an adverse event. In the long-term 6-month study in adult insomnia patients, 7.2% of 195 patients who received placebo and 12.8% of 593 patients who received 3 mg LUNESTA discontinued due to an adverse event. No event that resulted in discontinuation occurred at a rate of greater than 2%.
Adverse Events Observed At An Incidence Of > 2% In Controlled Trials
Table 1 shows the incidence of treatment-emergent adverse events from a Phase 3 placebo-controlled study of LUNESTA at doses of 2 or 3 mg in non-elderly adults. Treatment duration in this trial was 44 days. The table includes only events that occurred in 2% or more of patients treated with LUNESTA 2 mg or 3 mg in which the incidence in patients treated with LUNESTA was greater than the incidence in placebo-treated patients.
14
Table 1: Incidence (%) of Treatment-Emergent
Adverse Events in a 6-Week
Placebo-Controlled Study in Non-Elderly Adults with LUNESTA(1)
|
Adverse Event |
|
Placebo |
|
LUNESTA 2 mg |
|
LUNESTA 3 mg |
|
|
Body as a Whole |
|
|
|
|
|
|
|
|
Headache |
|
13 |
|
21 |
|
17 |
|
|
Viral Infection |
|
1 |
|
3 |
|
3 |
|
|
Digestive System |
|
|
|
|
|
|
|
|
Dry Mouth |
|
3 |
|
5 |
|
7 |
|
|
Dyspepsia |
|
4 |
|
4 |
|
5 |
|
|
Nausea |
|
4 |
|
5 |
|
4 |
|
|
Vomiting |
|
1 |
|
3 |
|
0 |
|
|
Nervous System |
|
|
|
|
|
|
|
|
Anxiety |
|
0 |
|
3 |
|
1 |
|
|
Confusion |
|
0 |
|
0 |
|
3 |
|
|
Depression |
|
0 |
|
4 |
|
1 |
|
|
Dizziness |
|
4 |
|
5 |
|
7 |
|
|
Hallucinations |
|
0 |
|
1 |
|
3 |
|
|
Libido Decreased |
|
0 |
|
0 |
|
3 |
|
|
Nervousness |
|
3 |
|
5 |
|
0 |
|
|
Somnolence |
|
3 |
|
10 |
|
8 |
|
|
Respiratory System |
|
|
|
|
|
|
|
|
Infection |
|
3 |
|
5 |
|
10 |
|
|
Skin and Appendages |
|
|
|
|
|
|
|
|
Rash |
|
1 |
|
3 |
|
4 |
|
|
Special Senses |
|
|
|
|
|
|
|
|
Unpleasant Taste |
|
3 |
|
17 |
|
34 |
|
|
Urogenital System |
|
|
|
|
|
|
|
|
Dysmenorrhea * |
|
0 |
|
3 |
|
0 |
|
|
Gynecomastia ** |
|
0 |
|
3 |
|
0 |
|
(1) Events for which the LUNESTA incidence was equal to or less than placebo are not listed on the table, but included the following: abnormal dreams, accidental injury, back pain, diarrhea, flu syndrome, myalgia, pain, pharyngitis, and rhinitis.
* Gender-specific adverse event in females
** Gender-specific adverse event in males
Adverse events from Table 1 that suggest a dose-response relationship in adults include viral infection, dry mouth, dizziness, hallucinations, infection, rash, and unpleasant taste, with this relationship clearest for unpleasant taste.
Table 2 shows the incidence of treatment-emergent adverse events from combined Phase 3 placebo-controlled studies of LUNESTA at doses of 1 or 2 mg in elderly adults (ages 65-86). Treatment duration in these trials was 14 days. The table includes only events that occurred in 2% or more of patients treated with LUNESTA 1 mg or 2 mg in which the incidence in patients treated with LUNESTA was greater than the incidence in placebo-treated patients.
15
Table 2: Incidence (%) of Treatment-Emergent Adverse
Events in Elderly
Adults (Ages 65-86) in 2-Week Placebo-Controlled Trials with LUNESTA(1)
|
Adverse Event |
|
Placebo |
|
LUNESTA 1 mg |
|
LUNESTA 2 mg |
|
|
Body as a Whole |
|
|
|
|
|
|
|
|
Accidental Injury |
|
1 |
|
0 |
|
3 |
|
|
Headache |
|
14 |
|
15 |
|
13 |
|
|
Pain |
|
2 |
|
4 |
|
5 |
|
|
Digestive System |
|
|
|
|
|
|
|
|
Diarrhea |
|
2 |
|
4 |
|
2 |
|
|
Dry Mouth |
|
2 |
|
3 |
|
7 |
|
|
Dyspepsia |
|
2 |
|
6 |
|
2 |
|
|
Nervous System |
|
|
|
|
|
|
|
|
Abnormal Dreams |
|
0 |
|
3 |
|
1 |
|
|
Dizziness |
|
2 |
|
1 |
|
6 |
|
|
Nervousness |
|
1 |
|
0 |
|
2 |
|
|
Neuralgia |
|
0 |
|
3 |
|
0 |
|
|
Skin and Appendages |
|
|
|
|
|
|
|
|
Pruritus |
|
1 |
|
4 |
|
1 |
|
|
Special Senses |
|
|
|
|
|
|
|
|
Unpleasant Taste |
|
0 |
|
8 |
|
12 |
|
|
Urogenital System |
|
|
|
|
|
|
|
|
Urinary Tract Infection |
|
0 |
|
3 |
|
0 |
|
(1) Events for which the LUNESTA incidence was equal to or less than placebo are not listed on the table, but included the following: abdominal pain, asthenia, nausea, rash, and somnolence.
Adverse events from Table 2 that suggest a dose-response relationship in elderly adults include pain, dry mouth, and unpleasant taste, with this relationship again clearest for unpleasant taste.
These figures cannot be used to predict the incidence of adverse events in the course of usual medical practice because patient characteristics and other factors may differ from those that prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses, and investigators. The cited figures, however, do provide the prescribing physician with some basis for estimating the relative contributions of drug and non-drug factors to the adverse event incidence rate in the population studied.
Other Events Observed During The Premarketing Evaluation Of LUNESTA
Following is a list of modified COSTART terms that reflect treatment-emergent adverse events as defined in the introduction to the ADVERSE REACTIONS section and reported by approximately 1550 subjects treated with LUNESTA at doses in the range of 1 to 3.5 mg/day during Phase 2 and 3 clinical trials throughout the United States and Canada. All reported events are included except those already listed in Tables 1 and 2 or elsewhere in labeling, minor events common in the general population, and events unlikely to be drug-related. Although the events reported occurred during treatment with LUNESTA, they were not necessarily caused by it.
16
Events are further categorized by body system and listed in order of decreasing frequency according to the following definitions: frequent adverse events are those that occurred on one or more occasions in at least 1/100 patients; infrequent adverse events are those that occurred in fewer than 1/100 patients but in at least 1/1,000 patients; rare adverse events are those that occurred in fewer than 1/1,000 patients. Gender-specific events are categorized based on their incidence for the appropriate gender.
Body as a Whole: Frequent: chest pain; Infrequent: allergic reaction, cellulitis, face edema, fever, halitosis, heat stroke, hernia, malaise, neck rigidity, photosensitivity.
Cardiovascular System: Frequent: migraine; Infrequent: hypertension; Rare: thrombophlebitis.
Digestive System: Infrequent: anorexia, cholelithiasis, increased appetite, melena, mouth ulceration, thirst, ulcerative stomatitis; Rare: colitis, dysphagia, gastritis, hepatitis, hepatomegaly, liver damage, stomach ulcer, stomatitis, tongue edema, rectal hemorrhage.
Hemic and Lymphatic System: Infrequent: anemia, lymphadenopathy.
Metabolic and Nutritional: Frequent: peripheral edema; Infrequent: hypercholesteremia, weight gain, weight loss; Rare: dehydration, gout, hyperlipemia, hypokalemia.
Musculoskeletal System: Infrequent: arthritis, bursitis, joint disorder (mainly swelling, stiffness, and pain), leg cramps, myasthenia, twitching; Rare: arthrosis, myopathy, ptosis.
Nervous System: Infrequent: agitation, apathy, ataxia, emotional lability, hostility, hypertonia, hypesthesia, incoordination, insomnia, memory impairment, neurosis, nystagmus, paresthesia, reflexes decreased, thinking abnormal (mainly difficulty concentrating), vertigo; Rare: abnormal gait, euphoria, hyperesthesia, hypokinesia, neuritis, neuropathy, stupor, tremor.
Respiratory System: Infrequent: asthma, bronchitis, dyspnea, epistaxis, hiccup, laryngitis.
Skin and Appendages: Infrequent: acne, alopecia, contact dermatitis, dry skin, eczema, skin discoloration, sweating, urticaria; Rare: erythema multiforme, furunculosis, herpes zoster, hirsutism, maculopapular rash, vesiculobullous rash.
Special Senses: Infrequent: conjunctivitis, dry eyes, ear pain, otitis externa, otitis media, tinnitus, vestibular disorder; Rare: hyperacusis, iritis, mydriasis, photophobia.
Urogenital System: Infrequent: amenorrhea, breast engorgement, breast enlargement, breast neoplasm, breast pain, cystitis, dysuria, female lactation, hematuria, kidney calculus, kidney pain, mastitis, menorrhagia, metrorrhagia, urinary frequency, urinary incontinence, uterine hemorrhage, vaginal hemorrhage, vaginitis; Rare: oliguria, pyelonephritis, urethritis.
17
DRUG ABUSE AND DEPENDENCE:
Controlled Substance Class
LUNESTA is a Schedule IV controlled substance under the Controlled Substances Act. Other substances under the same classification are benzodiazepines and the nonbenzodiazepine hypnotics zaleplon and zolpidem. While eszopiclone is a hypnotic agent with a chemical structure unrelated to benzodiazepines, it shares some of the pharmacologic properties of the benzodiazepines.
Abuse, Dependence, And Tolerance
Abuse And Dependence
Abuse and addiction are separate and distinct from physical dependence and tolerance. Abuse is characterized by misuse of the drug for non-medical purposes, often in combination with other psychoactive substances. Physical dependence is a state of adaptation that is manifested by a specific withdrawal syndrome that can be produced by abrupt cessation, rapid dose reduction, decreasing blood level of the drug and/or administration of an antagonist. Tolerance is a state of adaptation in which exposure to a drug induces changes that result in a diminution of one or more of the drug’s effects over time. Tolerance may occur to both the desired and undesired effects of drugs and may develop at different rates for different effects.
Addiction is a primary, chronic, neurobiological disease with genetic, psychosocial, and environmental factors influencing its development and manifestations. It is characterized by behaviors that include one or more of the following: impaired control over drug use, compulsive use, continued use despite harm, and craving. Drug addiction is a treatable disease, utilizing a multidisciplinary approach, but relapse is common.
In a study of abuse liability conducted in individuals with known histories of benzodiazepine abuse, eszopiclone at doses of 6 and 12 mg produced euphoric effects similar to those of diazepam 20 mg. In this study, at doses 2-fold or greater than the maximum recommended doses, a dose-related increase in reports of amnesia and hallucinations was observed for both LUNESTA and diazepam.
The clinical trial experience with LUNESTA revealed no evidence of a serious withdrawal syndrome. Nevertheless, the following adverse events included in DSM-IV criteria for uncomplicated sedative/hypnotic withdrawal were reported during clinical trials following placebo substitution occurring within 48 hours following the last LUNESTA treatment: anxiety, abnormal dreams, nausea, and upset stomach. These reported adverse events occurred at an incidence of 2% or less. Use of benzodiazepines and similar agents may lead to physical and psychological dependence. The risk of abuse and dependence increases with the dose and duration of treatment and concomitant use of other psychoactive drugs. The risk is also greater for patients who have a history of alcohol or drug abuse or history of psychiatric disorders. These patients should be under careful surveillance when receiving LUNESTA or any other hypnotic.
18
Tolerance
Some loss of efficacy to the hypnotic effect of benzodiazepines and benzodiazepine-like agents may develop after repeated use of these drugs for a few weeks.
No development of tolerance to any parameter of sleep measurement was observed over six months. Tolerance to the efficacy of LUNESTA 3 mg was assessed by 4-week objective and 6-week subjective measurements of time to sleep onset and sleep maintenance for LUNESTA in a placebo-controlled 44-day study, and by subjective assessments of time to sleep onset and WASO in a placebo-controlled study for 6 months.
OVERDOSAGE:
There is limited premarketing clinical experience with the effects of an overdosage of LUNESTA. In clinical trials with eszopiclone, one case of overdose with up to 36 mg of eszopiclone was reported in which the subject fully recovered. Individuals have fully recovered from racemic zopiclone overdoses up to 340 mg (56 times the maximum recommended dose of eszopiclone).
Signs And Symptoms
Signs and symptoms of overdose effects of CNS depressants can be expected to present as exaggerations of the pharmacological effects noted in preclinical testing. Impairment of consciousness ranging from somnolence to coma has been described. Rare individual instances of fatal outcomes following overdose with racemic zopiclone have been reported in European postmarketing reports, most often associated with overdose with other CNS-depressant agents.
Recommended Treatment
General symptomatic and supportive measures should be used along with immediate gastric lavage where appropriate. Intravenous fluids should be administered as needed. Flumazenil may be useful. As in all cases of drug overdose, respiration, pulse, blood pressure, and other appropriate signs should be monitored and general supportive measures employed. Hypotension and CNS depression should be monitored and treated by appropriate medical intervention. The value of dialysis in the treatment of overdosage has not been determined.
Poison Control Center
As with the management of all overdosage, the possibility of multiple drug ingestion should be considered. The physician may wish to consider contacting a poison control center for up-to-date information on the management of hypnotic drug product overdosage.
19
DOSAGE AND ADMINISTRATION:
The dose of LUNESTA should be individualized. The recommended starting dose for LUNESTA for most non-elderly adults is 2 mg immediately before bedtime. Dosing can be initiated at or raised to 3 mg if clinically indicated, since 3 mg is more effective for sleep maintenance (see Precautions).
The recommended starting dose of LUNESTA for elderly patients whose primary complaint is difficulty falling asleep is 1 mg immediately before bedtime. In these patients, the dose may be increased to 2 mg if clinically indicated. For elderly patients whose primary complaint is difficulty staying asleep, the recommended dose is 2 mg immediately before bedtime (see Precautions).
Taking LUNESTA with or immediately after a heavy, high-fat meal results in slower absorption and would be expected to reduce the effect of LUNESTA on sleep latency (see Pharmacokinetics under Clinical Pharmacology).
Special Populations
Hepatic
The starting dose of LUNESTA should be 1 mg in patients with severe hepatic impairment. LUNESTA should be used with caution in these patients.
Coadministration With CYP3A4 Inhibitors
The starting dose of LUNESTA should not exceed 1 mg in patients coadministered LUNESTA with potent CYP3A4 inhibitors. If needed, the dose can be raised to 2 mg.
HOW SUPPLIED:
LUNESTA 3 mg tablets are round, dark blue, film-coated, and identified with debossed markings of S193 on one side, and are supplied as:
|
NDC 00000-000-00 |
|
bottle of 100 tablets |
|
NDC 00000-000-00 |
|
carton of 90 tablets |
LUNESTA 2 mg tablets are round, white, film-coated, and identified with debossed markings of S191 on one side, and are supplied as:
|
NDC 00000-000-00 |
|
bottle of 100 tablets |
|
NDC 00000-000-00 |
|
carton of 90 tablets |
LUNESTA 1 mg tablets are round, light blue, film-coated, and identified with debossed markings of S190 on one side, and are supplied as:
20
|
NDC 00000-000-00 |
|
bottle of 100 tablets |
Store at 25°C (77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
21

Manufactured for:
Sepracor Inc.
Xxxxxxxxxxx, XX 00000 XXX
by Xxxxxxx Xxx., Xxxxxxxxxxx, Xxxxxxx X0X 0X0 Xxxxxx
For customer service, call 0-000-000-0000.
To report adverse events, call 0-000-000-0000.
For medical information, call 0-000-000-0000.
April 2007
PHARMACIST — DETACH HERE AND GIVE INFORMATION TO PATIENT.
|
Rx only |
|
C-IV |
LUNESTA® (eszopiclone) TABLETS
1 mg, 2 mg, 3 mg
INFORMATION FOR PATIENTS TAKING LUNESTA
Your doctor has prescribed LUNESTA to help you sleep. The following information is intended to guide you in the safe use of this medicine. It is not meant to take the place of your doctor’s instructions. If you have any questions about LUNESTA tablets, be sure to ask your doctor or pharmacist.
LUNESTA is used to treat different types of sleep problems, such as difficulty in falling asleep, difficulty in maintaining sleep during the night, and waking up too early in the morning. Most people with insomnia have more than one of these problems. You should take LUNESTA immediately before going to bed because of the risk of falling.
LUNESTA belongs to a group of medicines known as “hypnotics” or, simply, sleep medicines. There are many different sleep medicines available to help people sleep better. Insomnia is often transient and intermittent. It usually requires treatment for only a short time, usually 7 to 10 days up to 2 weeks. Some people have chronic sleep problems that may require more prolonged use of sleep medicine. However, you should not use these medicines for long periods without talking with your doctor about the risks and benefits of prolonged use.
22
Side Effects
All medicines have side effects. The most common side effects of sleep medicines are:
· Drowsiness
· Dizziness
· Lightheadedness
· Difficulty with coordination
Sleep medicines can make you sleepy during the day. How drowsy you feel depends upon how your body reacts to the medicine, which sleep medicine you are taking, and how large a dose your doctor has prescribed. Daytime drowsiness is best avoided by taking the lowest dose possible that will still help you sleep at night. Your doctor will work with you to find the dose of LUNESTA that is best for you. Some patients taking LUNESTA have reported next-day sleepiness.
To manage these side effects while you are taking this medicine:
· When you first start taking LUNESTA or any other sleep medicine, until you know whether the medicine will still have some effect on you the next day, use extreme care while doing anything that requires complete alertness, such as driving a car, operating machinery, or piloting an aircraft.
· Do not drink alcohol when you are taking LUNESTA or any sleep medicine. Alcohol can increase the side effects of LUNESTA or any other sleep medicine.
· Do not take any other medicines without asking your doctor first. This includes medicines you can buy without a prescription. Some medicines can cause drowsiness and are best avoided while taking LUNESTA.
· Always take the exact dose of LUNESTA prescribed by your doctor. Never change your dose without talking to your doctor first.
Special Concerns
There are some special problems that may occur while taking sleep medicines.
Memory Problems
Sleep medicines may cause a special type of memory loss or “amnesia.” When this occurs, a person may not remember what has happened for several hours after taking the medicine. This is usually not a problem since most people fall asleep after taking the medicine. Memory loss can be a problem, however, when sleep medicines are taken while traveling, such as during an airplane flight and the person wakes up before the effect of the medicine is gone. This has been called “traveler’s amnesia.” Memory problems have been reported rarely by patients taking LUNESTA in clinical studies. In most cases, memory problems can be avoided if you take
23
LUNESTA only when you are able to get a full night of sleep before you need to be active again. Be sure to talk to your doctor if you think you are having memory problems.
Tolerance
When sleep medicines are used every night for more than a few weeks, they may lose their effectiveness in helping you sleep. This is known as “tolerance.” Development of tolerance to LUNESTA was not observed in a clinical study of 6 months’ duration. Insomnia is often transient and intermittent, and prolonged use of sleep medicines is generally not necessary. Some people, though, have chronic sleep problems that may require more prolonged use of sleep medicine. If your sleep problems continue, consult your doctor, who will determine whether other measures are needed to overcome your sleep problems.
Dependence
Sleep medicines can cause dependence in some people, especially when these medicines are used regularly for longer than a few weeks or at high doses. Dependence is the need to continue taking a medicine because stopping it is unpleasant.
When people develop dependence, stopping the medicine suddenly may cause unpleasant symptoms (see Withdrawal below). They may find they have to keep taking the medicine either at the prescribed dose or at increasing doses just to avoid withdrawal symptoms.
All people taking sleep medicines have some risk of becoming dependent on the medicine. However, people who have been dependent on alcohol or other drugs in the past may have a higher chance of becoming addicted to sleep medicines. This possibility must be considered before using these medicines for more than a few weeks. If you have been addicted to alcohol or drugs in the past, it is important to tell your doctor before starting LUNESTA or any sleep medicine.
Withdrawal
Withdrawal symptoms may occur when sleep medicines are stopped suddenly after being used daily for a long time. In some cases, these symptoms can occur even if the medicine has been used for only a week or two. In mild cases, withdrawal symptoms may include unpleasant feelings. In more severe cases, abdominal and muscle cramps, vomiting, sweating, shakiness, and, rarely, seizures may occur. These more severe withdrawal symptoms are very uncommon. Although withdrawal symptoms have not been observed in the relatively limited controlled trials experience with LUNESTA, there is, nevertheless, the risk of such events in association with the use of any sleep medicine.
Another problem that may occur when sleep medicines are stopped is known as “rebound insomnia.” This means that a person may have more trouble sleeping the first few nights after the medicine is stopped than before starting the medicine. If you should experience rebound insomnia, do not get discouraged. This problem usually goes away on its own after 1 or 2 nights.
24
If you have been taking LUNESTA or any other sleep medicine for more than 1 or 2 weeks, do not stop taking it on your own. Always follow your doctor’s directions.
Changes In Behavior And Thinking
Some people using sleep medicines have experienced unusual changes in their thinking and/or behavior. These effects are not common. However, they have included:
· More outgoing or aggressive behavior than normal
· Confusion
· Strange behavior
· Agitation
· Hallucinations
· Worsening of depression
· Suicidal thoughts
How often these effects occur depends on several factors, such as a person’s general health, the use of other medicines, and which sleep medicine is being used. Clinical experience with LUNESTA suggests that it is rarely associated with these behavior changes.
It is also important to realize that it is rarely clear whether these behavior changes are caused by the medicine, are caused by an illness, or have occurred on their own. In fact, sleep problems that do not improve may be due to illnesses that were present before the medicine was used. If you or your family notice any changes in your behavior, or if you have any unusual or disturbing thoughts, call your doctor immediately.
Pregnancy And Breastfeeding
Sleep medicines may cause sedation or other potential effects in the unborn baby when used during the last weeks of pregnancy. Be sure to tell your doctor if you are pregnant, if you are planning to become pregnant, or if you become pregnant while taking LUNESTA.
In addition, a very small amount of LUNESTA may be present in breast milk after use of the medication. The effects of very small amounts of LUNESTA on an infant are not known; therefore, as with all other prescription sleep medicines, it is recommended that you not take LUNESTA if you are breastfeeding a baby.
Safe Use Of Sleep Medicines
To ensure the safe and effective use of LUNESTA or any other sleep medicine, you should observe the following cautions:
1. LUNESTA is a prescription medicine and should be used ONLY as directed by your doctor. Follow your doctor’s instructions about how to take, when to take, and how long to take LUNESTA.
25
2. Never use LUNESTA or any other sleep medicine for longer than directed by your doctor.
3. If you notice any unusual and/or disturbing thoughts or behavior during treatment with LUNESTA or any other sleep medicine, contact your doctor.
4. Tell your doctor about any medicines you may be taking, including medicines you may buy without a prescription and herbal preparations. You should also tell your doctor if you drink alcohol. DO NOT use alcohol while taking LUNESTA or any other sleep medicine.
5. Do not take LUNESTA unless you are able to get 8 or more hours of sleep before you must be active again.
6. Do not increase the prescribed dose of LUNESTA or any other sleep medicine unless instructed by your doctor.
7. When you first start taking LUNESTA or any other sleep medicine, until you know whether the medicine will still have some effect on you the next day, use extreme care while doing anything that requires complete alertness, such as driving a car, operating machinery, or piloting an aircraft.
8. Be aware that you may have more sleeping problems the first night or two after stopping any sleep medicine.
9. Be sure to tell your doctor if you are pregnant, if you are planning to become pregnant, if you become pregnant, or if you are breastfeeding a baby while taking LUNESTA.
10. As with all prescription medicines, never share LUNESTA or any other sleep medicine with anyone else. Always store LUNESTA or any other sleep medicine in the original container and out of reach of children.
11. Be sure to tell your doctor if you suffer from depression.
12. LUNESTA works very quickly. You should only take LUNESTA immediately before going to bed.
13. For LUNESTA to work best, you should not take it with or immediately after a high-fat, heavy meal.
14. Some people, such as older adults (i.e., ages 65 and over) and people with liver disease, should start with the lower dose (1 mg) of LUNESTA. Your doctor may choose to start therapy at 2 mg. In general, adults under age 65 should be treated with 2 or 3 mg.
15. Each tablet is a single dose; do not crush or break the tablet.
26
Manufactured for:
Sepracor Inc.
Xxxxxxxxxxx, XX 00000 XXX
by Xxxxxxx Xxx., Xxxxxxxxxxx, Xxxxxxx X0X 0X0 Xxxxxx
For customer service, call 0-000-000-0000.
To report adverse events, call 0-000-000-0000.
For medical information, call 0-000-000-0000.
April 2007
27
CONFIDENTIAL
SCHEDULE 7.3(c)
Hypothetical Royalty Calculation
Hypothetical Scenario: In the third Calendar Year following first launch in the GSK Territory, the following Net Sales were achieved by GSK, its Affiliates and Sublicensees:
• Net Sales within countries within the GSK Territory where in each country sales of Generic Products exceed [**] percent ([**]%) of the total sales of Product by GSK, its Affiliates and Sublicensees in such country (“Generic Countries”):
|
Q1 |
|
[**] |
|
Q2 |
|
[**] |
|
Q3 |
|
[**] |
|
Q4 |
|
[**] |
• Net Sales within countries within the GSK Territory where there are no Generic Products or sales of Generic Products are less than [**] percent ([**]%) of the total sales of Product by GSK, its Affiliates and Sublicensees in such country (“Non-Generic Countries”):
|
Q1 |
|
[**] |
|
Q2 |
|
[**] |
|
Q3 |
|
[**] |
|
Q4 |
|
[**] |
• Total Net Sales within the GSK Territory:
|
Q1 |
|
[**] |
|
Q2 |
|
[**] |
|
Q3 |
|
[**] |
|
Q4 |
|
[**] |
Royalty calculation:
Q1 Total sales in Non-Generic Countries is US$[**]. Royalty rate applicable in those countries during Q1 is therefore [**]%.
Royalties payable in Q1 for Non-Generic Countries = [**]
Total sales in Generic Countries is US$[**]. As cumulative year to date sales in Non-Generic Countries is less than US$[**], royalty rate applicable in Generic Countries during Q1 is [**]%.
Royalties payable in Q1 for Generic Countries = [**]
Q2 Cumulative year to date sales in all Non-Generic Countries is US$[**]. Of the US$[**] sales occurring in Q2, US$[**] will attract the [**]% royalty rate and US$[**] will attract the [**]% royalty rate.
Royalties payable in Q2 for Non-Generic Countries = [**].
Royalties payable in Generic Countries will payable in same proportions, applied to the sales occurring in Q2,
i.e [**]
Royalties payable in Q2 for Generic Countries = [**]
CONFIDENTIAL
Q3 Cumulative year to date sales in all Non-Generic Countries is US$[**]. Of the US$[**] sales occurring in Q3, US$[**] will attract the [**]% royalty rate, and US$[**] will attract the [**]% royalty rate.
Royalties payable in Q3 for Non-Generic Countries = [**].
Royalties payable in Generic Countries will payable in same proportions, applied to the sales occurring in Q3, i.e [**]
Royalties payable in Q3 for Generic Countries = [**]
Q4 Cumulative year to date sales in all Non-Generic Countries is US$[**]. Of the US$[**] sales occurring in Q4, all of the US$[**] will attract the [**]% royalty rate.
Royalties payable in Q4 for Non-Generic Countries = [**].
Royalties payable in Generic Countries will payable in same proportions, applied to sales occurring in Q4, i.e. [**]% will be payable at [**]%.
Royalties payable in Q4 for Generic Countries = [**]
CONFIDENTIAL
SCHEDULE 7.3(e)
Hypothetical Calculation of Cost of Bulk Tablets and Net Sales
In a hypothetical Calendar Year:
(A) the average price invoiced during such Calendar Year for the supply by Sepracor to GSK of a [**]mg Bulk Tablets of Product was US$[**] and
(B) the number of [**]mg tablets of Product sold by GSK, its Affiliates or Sub-licensees during such Calendar Year was [**].
The aggregate price paid by GSK for [**]mg Bulk Tablets for Product sold in such Calendar Year is therefore A x B = US$[**]
A similar calculation for other dosage forms, meant that in this hypothetical Calendar Year, the aggregate price paid by GSK for Bulk Tablets in all forms for Product sold in such Calendar Year was, for the sake of this example, US$[**].
If the Net Sales in that Calendar Year were US$[**], and the royalties payable on such Net Sales were US$[**], the calculation required under Section 7.3(e) would be: [**]
In this example, the [**]% cap would not be reached, and there would be no adjustment made under Section 7.3(e).
If however, the aggregate price paid by GSK for Bulk Tablets in all forms for Product sold in such Calendar Year had been, for the sake of this example, US$[**], the calculation required under Section 7.3(e) would be: [**]
In this case, as [**]% of US$[**] is US$[**], an adjustment of US$[**] would need to be made to the royalties paid for such Calendar Year [**]
CONFIDENTIAL
SCHEDULE 10.2
TERM SHEET FOR MANUFACTURING AND SUPPLY
1. Definitive Agreement. Following execution of the Development, License and Commercialization Agreement between Sepracor and GSK (“License Agreement”), Sepracor and GSK shall, in good faith, negotiate and enter into a manufacturing and supply agreement that incorporates, among other things, the terms and conditions set forth in this term sheet (the “Supply Agreement”). Capitalized terms used but not otherwise defined herein shall have the meaning ascribed to such terms in the License Agreement.
2. Supply of Products. Sepracor shall manufacture or have manufactured exclusively for GSK’s use in the GSK Territory Bulk Tablets for commercial sale (including physician samples) and Clinical Supplies. Based on the Forecast as described herein, Sepracor shall maintain at all times sufficient capacity to meet all GSK’s, GSK’s Affiliates’ and GSK’s Sublicensees’ requirements for Bulk Tablets to enable them to meet their respective needs for Product for the GSK Territory.
3. Product Requirements. All Bulk Tablets supplied by Sepracor will be manufactured in accordance with the relevant standards of cGMP and applicable laws and regulations (both at the site of manufacture and where the Bulk Tablets are to be supplied) and will comply with the relevant specifications, each Marketing Approval, and will be fit for purpose. The parties recognize that they will need to agree upon specifics of manufacturing requirements for individual countries or regulatory areas either in advance of, or as part of, the Marketing Approval process.
4. Price. The purchase price of Bulk Tablets for commercial supplies shall be Sepracor’s [**], and for Clinical Supplies and physician samples shall be [**]. For the avoidance of doubt, in any country of the GSK Territory where market pricing does not allow for GSK to profitably sell the Product with Bulk Tablets being purchased at this rate, Sepracor is under no obligation to supply Product at a purchase price lower than as stated above. Subject to GSK agreeing confidentiality terms in the Supply Agreement, on terms no less onerous than those contained in Section 11 of the License Agreement, the parties will operate on [**] (with associated audit rights) and shall meet regularly (and no less than once each calendar year) to agree the [**] to be invoiced to GSK as well as the division between Clinical Supplies, physician samples and commercial supplies. The Supply Agreement will also provide for initiatives to manage costs and identify potential areas for cost reduction as part of a continuous improvement programme.
5. Forecasts and Firm Orders. Upon the execution of the Supply Agreement and [**] thereafter, GSK shall provide Sepracor with a rolling [**] forecast of its estimated quarterly purchases of Bulk Tablets (the “Forecast”) for both commercial and Clinical
CONFIDENTIAL
Supply needs. Except as set forth below, each Forecast will be non-binding and will represent only GSK’s good faith estimate of expected orders for the Bulk Tablets. However, for each such Forecast, the [**] will be a firm order (purchase orders for such firm orders will also be provided in accordance with Supply Agreement) and will represent GSK’s commitment to Sepracor to purchase the amount of Bulk Tablets indicated in such portion of the Forecast. For the [**] period of each such Forecast, the quantities of Bulk Tablets actually purchased by GSK shall not be [**]. GSK shall also provide Sepracor with non-binding mid-term and long-term forecasts on a yearly basis (for years [**]), which shall not exceed [**] years.
6. Shipping and Delivery. The delivery terms in the Supply Agreement shall be Ex Works (Incoterms 2000). Risk of loss and title shall transfer at the time of delivery of Bulk Tablets to the carrier. Each delivery will be accompanied by the corresponding Certificate of Analysis and relevant delivery documentation.
7. Packaging and Labeling. GSK will package (including label) (or have packaged and labelled) the Bulk Tablets for commercial sale (including physician samples) and Clinical Supplies. Bulk Tablets supplied to GSK will be appropriately packaged and labeled based upon specifications agreed between the Parties and in accordance with all applicable laws and regulations.
8. Acceptance and Rejection. GSK shall have [**] from its receipt of the Bulk Tablets at GSK’s designated facility to notify Sepracor of its rejection of the Bulk Tablets for any defects which are apparent from carrying out incoming inspection and control procedures (to be further defined in the Quality Agreement). This shall be without prejudice to rights and obligations of the Parties in respect of latent defects. The Supply Agreement shall include provisions for the referral of disputes over alleged defects in Bulk Tablets to an independent laboratory for resolution.
9. Payment. Sepracor shall invoice GSK upon shipment of each released batch of Bulk Tablets. Payment terms will be thirty (30) days from date of receipt of the Bulk Tablets (with a paper copy of the related invoice being sent electronically and by post). Any payments or portions thereof due under this Agreement that are not paid by the date such payment is due shall bear interest at the rate set forth in Section 8.5 of the License Agreement.
10. Term. The Supply Agreement shall be coterminous with the License Agreement, on a country-by-country basis.
11. Inability to Supply. Sepracor will maintain an inventory of API equivalent to [**] forward cover of Bulk Tablets (or such other inventory as mutually agreed) (“Reserve Supply”), which shall be used to meet any disruption or shortfall in supply that may occur for any reason during the term of the Supply Agreement.
In the event of any Inability to Supply, (or any anticipated Inability to Supply) Sepracor shall notify GSK immediately and shall cooperate with GSK in taking at Sepracor’s cost (except where the Inability to Supply is due to an event of force majeure, in which case the parties will agree an equitable allocation of the costs) all actions that are reasonably necessary in order to remedy such Inability to Supply, including making up the shortfall from the Reserve Supply.
An “Inability to Supply” shall mean Sepracor’s failure for any reason, including without limitation force majeure or otherwise, to supply GSK with at least, in the aggregate, [**] percent ([**]%) of the quantities of the Bulk Tablets Forms ordered by GSK for any [**] period.
In the event of any Inability to Supply, the Parties will review through the JSC the amount of Bulk Tablets available and if necessary will agree appropriate rationing of available supply as between the Sepracor Territory and the GSK Territory to ensure that supply to GSK is not disproportionately disadvantaged.
In the event of an Inability to Supply where Sepracor is unable to make up the shortfall from the Reserve Supply or otherwise by way of product rationing from the available supply, upon written request by GSK, Sepracor shall provide GSK with such assistance (other than financial assistance) and any know-how Controlled by Sepracor, as reasonably necessary for manufacturing Bulk Tablets Forms including, without limitation, setting up a manufacturing facility at GSK. In connection with the foregoing, GSK shall be permitted to consult with Sepracor’s technical personnel, and such technical personnel will provide such assistance as may be necessary to transfer the specified manufacturing activities and relevant Sepracor Know-How to GSK (or its nominated contract manufacturer) and Sepracor shall immediately grant GSK a limited, non-exclusive royalty free license under the Sepracor Technology for the sole purpose of manufacturing (or having manufactured) Bulk Tablets in or outside the GSK Territory for packaging into Product for commercial sale (including physician samples) and Clinical Supplies in the GSK Territory, and with the agreement of GSK make any necessary consequential amendments to the License Agreement. Sepracor shall be responsible for Sepracor’s actual costs and expenses incurred in connection with such technology transfer.
The remedies set out in this paragraph 11 shall be without prejudice to any other rights and remedies under the Supply Agreement for failure to meet firm orders for Bulk Tablets.
12. Quality Agreement and Pharmacovigilance Agreement. In addition to the execution of the Supply Agreement, the Parties will enter into a Quality Agreement and a Pharmacovigilance Agreement incorporating terms required in the License Agreement and such other terms appropriate for manufacture of Bulk Tablets for commercial distribution (including physician samples) and Clinical Supplies.
CONFIDENTIAL
13. Pre-Supply Approval. GSK will have the right to conduct pre-supply approval audits of those manufacturing facilities of Sepracor, and any appointed third party manufacturer, which are used in the manufacture of Bulk Tablets for GSK, prior to taking supply. Thereafter, Sepracor shall not appoint any other third party manufacturer nor change the location of any part of the manufacturing process for the manufacture of Bulk Tablets for GSK without GSK’s prior written consent, not to be unreasonably withheld, and subject to the change control procedure set out in the Quality Agreement.
14. Audits & Inspections. GSK shall have the right to carry out, not more than annually (except for “for cause” audits), upon reasonable prior notice (or at any time for cause), regular GMP, EHS and loss prevention audits of those parts of Sepracor’s manufacturing site(s) used in the manufacture of Bulk Tablets for GSK. Sepracor shall use Commercially Reasonable Efforts to procure the same rights for GSK at the premises of its third party manufacturers. If Sepracor is unable to procure such rights, Sepracor shall carry out such audits and inspections itself on behalf of GSK and shall report the results to GSK. Where any audit identifies work to be completed to ensure cGMP compliance or to meet the requirements of the Supply Agreement or the Quality Agreement, the cost of such activity will be borne by Sepracor.
15. Other Standard Terms. The Supply Agreement will also contain other provisions customary for agreements of this nature addressing the effect of termination, regulatory inspections, supply performance, quality assurance and assurance of supply, representations and warranties, indemnification, insurance and confidentiality and such other terms as may be necessary to reflect GSK or Sepracor corporate policies (including GSK’s policy on Ethics & Human Rights).
CONFIDENTIAL
SCHEDULE 16.2(g)
THREATENED AND PENDING LITIGATION IN GSK TERRITORY
None